New Human Physiology | Paulev-Zubieta 2nd Edition
Chapter 27 : Blood Glucose and Diabetes
| HOME | PREFACE | TABLE OF CONTENTS | SYMBOLS | SECTION INFO | CONTRIBUTORS | LINKS | CONTACT US |
Highlights
Study_ObjectivesPrinciplesDefinitionsEssentials
PathophysiologyEquationsSelf-AssessmentAnswers
Further Reading
|
Chapter 27
|
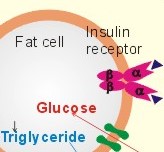 |
|
|
· To define glicentin, gluconeogenesis, glycogenolysis, glycolysis, hyperglycaemia, hypoglycaemia, incretins, insulin antagonists, paracrine secretion, primary and secondary diabetes mellitus. · To describe the structural and functional characteristics of the Langerhans islets with cell types and hormones produced. To describe the incretin effect, insulin effects, the insulin receptor, and the glucose transporter. To describe disorders of the different cell types and their clinical picture. To describe methods for evaluation of the glucose combustion. · To draw oral and intravenous glucose tolerance curves with linear and logarithmic ordinates for blood glucose concentration. · To explain the biosynthesis and the effects of insulin, glucagon, pancreatic polypeptide (PP) and somatostatin. To explain the glucose metabolism and the control of blood glucose in the fed and the fasting state. To explain the consequences and the therapy of high and low blood glucose disorders. · To use the above concepts in problem solving and in case histories. · The physiological principle in treatment of diabetes is to inject a fast-acting insulin three times a day just before meals and a slow-acting insulin at night. · Insulin promotes the storage of energy, the synthesis of glycogen, mRNA and proteins. · Certain major tissues (kidney, brain and intestine) are insensitive to the direct action of insulin. · Glicentin is intestinal glucagon. Glicentin is built up from 69 amino acids in contrast to pancreatic glucagon, which consists of 29 amino acid moieties. · Gluconeogenesis refers to formation of new glucose from glycogenic amino acids, lactate, glycerol and pyruvate. · Glycogenolysis refers to glycogen breakdown to glucose in the liver. · Glycolysis refers to anaerobic breakdown of glycogen. · Hyperglycaemia is a condition, where the blood glucose is above 6.7 mM. · Hypoglycaemia refers to a serious condition, where the blood glucose is below 2 mM. · Incretins are hormones, which strongly potentiates the insulin secretion induced by the rising blood [glucose]. The incretins cause a much larger insulin secretion than the iv. administration of glucose, even at the same rise in blood [glucose]. This extra insulin secretion is called the incretin effect. · Ketogenesis refers to accelerated lipolysis with liberation of free fatty acids to the blood. Free fatty acids are broken down to fatty acyl carnitine within the liver cells, and this molecule is converted into acetyl CoA, which in turn reach the mitochondria, where ketone bodies are formed. · Paracrine secretion is a release of signal molecules to neighbour cells. · Insulin antagonists are hormones opposing the effect of insulin: Pancreatic and intestinal (glicentin) glucagon, ACTH, growth hormone. · Primary diabetes mellitus refers to all cases, where the cause is not fully explained. · Secondary diabetes mellitus is caused by hypersecretion of one or more of the many catabolic hormones with hyperglycaemic effect (adrenaline, noradrenaline, glucagon, glucocorticoids and growth hormone) or by total destruction of the pancreas from pancreatitis or carcinoma. The hormone disorders are phaeochromocytoma, glucagonoma, Cushing’s syndrome and acromegaly. · Somatostatin (GHIH) is a multipotent hormone inhibitor consisting of a disulphide bridge and 14 amino acid units. This paragraph covers 1. the blood glucose regulation in the fed state, as well as in 2. the fasting states. Also 3. The endocrine pancreas, and 4. Pancreatic exocrine control is dealt with. 1. Glucose regulation in the fed state In the absorptive state after a balanced meal, nutrients enter the blood and lymph from the gastrointestinal tract (as monosaccharides, triglycerides, and amino acids). All the blood passes directly to the liver, which converts most of the other monosaccharides into glucose. Much of the absorbed carbohydrate enters the liver cells, but little of it is oxidised; instead most is stored as glycogen. Absorbed glucose, which did not enter hepatocytes but remained in the blood, is stored as glycogen by muscle cells, or it may enter into adipose tissue. A large fraction is oxidised to CO2 and water in the various cells of the body. Glucose is the major source of energy during the absorptive state. Homeostatic mechanisms maintain the plasma [glucose] within narrow limits in healthy humans, so that the energy needs during the postabsorptive state can be met by stored fuel. A high glucose intake results in a high blood [glucose] or extracellular hyperglycaemia. Hyperglycaemia increases insulin secretion from the b-cells and inhibits glucagon secretion from the a-cells of the pancreatic islets. These hormones block hepatic glucose production by glycogenolysis and gluconeogenesis. Insulin secretion dominates over all insulin-antagonists (growth hormone, glucagon, cortisol and some catecholamines). The sight and the smell of a meal triggers cephalic insulin secretion. When the meal reaches the intestine, several peptides of the incretin family are released; this is the intestinal secretion phase. Typical representatives of the incretin family are Gastric Inhibitory Peptide (GIP), glicentin (intestinal glucagon), and glucagon-like peptides (GLP-1 and -2). Incretins strongly potentate the insulin secretion induced by the rising blood [glucose]. The incretins cause a much larger insulin secretion than the iv. administration of glucose, even at the same rise in blood [glucose]. This extra insulin secretion is called the incretin effect. The insulin released following a meal increases the storage rate of glucose-related energy in the liver, muscles and fat tissues. The storage effect is much larger than when glucose is administered intravenously. Glucose is absorbed through the luminal membrane of the intestinal cells in glucose-Na+ transporter proteins. The two substances pass through the basolateral membrane via separate routes: Glucose passes in a special glucose-transporter, and Na+ is transferred by the Na+-K+‑pump. Glucose transport proteins and insulin receptors are described in Chapter 1. The filtration flux for glucose (mmol per min) increases proportionally to the concentration in the blood (as for all other filtered substances). Normally, all glucose is reabsorbed in the first part of the proximal renal tubules with a Tmax of 1.8 mmol per min or 320 mg per min. In other words, the passage fraction falls from one to zero already halfway through the proximal tubules. The excretion flux for glucose is zero in healthy humans. Glucose appears in the urine of diabetics, who have a blood [glucose] exceeding the appearance threshold (10 mM). Reabsorption of glucose over the luminal membrane of the proximal tubule cell takes place through the glucose-Na+ transporter. 2. Glucose regulation in the fasting state (rest and exercise) We keep our blood [glucose] surprisingly constant around the fasting level, considering the wide variety of daily activities. Glucose production (gluconeogenesis and glycogenolysis) must equal glucose combustion in the fasting state. Thus, a precise relation must exist between the secretion of insulin and glucagon from the pancreatic islets. In the fasting state hepatic glycogenolysis produces most of the glucose and the remaining glucose is produced by gluconeogenesis. Hepatic glucose is produced by glycogenolysis (glycogen breakdown to glucose) and by gluconeogenesis (glucose formation from glycogenic amino acids, lactate, glycerol, and small amounts of pyruvate. Muscle glycogen cannot deliver glucose to the blood, since muscle tissues lack glucose-6-phosphatase. In the fasting condition a healthy adult has a blood [glucose] of 4.5-5.6 mM. With an average [glucose] of 4 mM in 15 l of extracellular fluid volume (ECV), therefore, the total glucose content in ECV is 60 mmol or 10.8 g (2 teaspoons of sugar). This amount is equal to the glucose eliminated in one hour at rest (60 mmol each hour), but during maximum exercise that same person may use five times more glucose. The CNS and the erythrocytes neither synthesise nor store glucose, which is their primary fuel. Any surplus of glucose is deposited as liver and muscle glycogen. The liver cells contain an especially efficient glucose transporter (GLUT 2), and its glucose uptake rate cannot be increased further by insulin or by other hormones. Any small fall in blood glucose releases more glucagon. During fasting glycogenolysis, gluconeogenesis, and lipolysis are dominant. If a normal person does not eat for 24-48 hours the CNS cells revert to combustion of ketone bodies, and a reversible condition that is similar to diabetes develops (hunger diabetes). Exercise The exercise stress on the hypothalamus increases the activity of the sympathoadrenergic system, which includes increased secretion of adrenaline from the adrenal medulla. Sympathoadrenergic activity inhibits the insulin secretion from the b-cells. Sympathetic activity also increases hepatic glucose production. We tend to increase glucagon secretion only if the blood [glucose] falls. A slight fall in blood [glucose] can occur both during exercise bouts and during prolonged exercise. During exercise the blood [glucose] is maintained rather constant by bihormonal interplay between insulin and glucagon. Generally, an increasing demand of more energy elicits increased glycogenolysis, lipolysis, and increased gluconeogenesis caused by insulin-antagonistic hormones (catecholamines, glucagon, cortisol, and growth hormone). Adrenaline inhibits the insulin and stimulates the glucagon secretion, so the blood [glucose] increases. Somatotropin ‑ human growth hormone (GH) ‑ is an insulin-antagonist, but together with insulin probably the most important anabolic hormone. Conditions where energy sources are lacking are hypoglycaemia, hunger, fasting state, exhaustion, and stress. These conditions trigger the release of GRH from the hypothalamus, which in turn stimulates the release of GH from the hypophysis. This hormone has a tropic effect on the a -cells of the pancreatic islets. GH releases glucagon from these cells, just as sympathetic stimulation from the hypothalamus does. GH increases blood [glucose] by increasing hepatic glucose production (glycogenolysis but not its gluconeogenesis) and by inhibiting the insulin sensitivity of the muscle cells and thus reduces their glucose uptake. GH also has the same effect on fat cells, mobilising fatty acids and glycerol. GH stimulates protein synthesis, mitoses, chondrogenesis, ossification, and phosphate balance, while increasing glycolysis (ie, anaerobic breakdown of glycogen). Glucocorticoids are insulin-antagonists. They stimulate the hepatic glucose production (glycogenolysis and gluconeogenesis) but inhibit the cellular glucose uptake. Glucocorticoids are permissive and potentiating for catecholamines and glucagon. Catecholamines (adrenaline & noradrenaline) are insulin-antagonists. Adrenaline stimulates hepatic glucose production (glycogenolysis). Catecholamines also stimulate lipolysis. The increase in mitochondrial oxygen uptake by T3/T4 is potentates by catecholamines. The glucostat Glucose sensitive neurons in the hypothalamus (the glucostatic centre) react to hypoglycaemia by releasing glucagon from the pancreatic a-cells and catecholamines from the adrenal medulla by action of the sympathetic system. The glucostatic centre also reacts to hyperglycaemia to release insulin from pancreatic b-cells, and to activate hepatic glycogen synthesis by vagal stimuli. Insulin promotes the entry of glucose into tissues, including the neurons of the hypothalamic glucostatic centre (but in no other brain neurons). A balanced blood [glucose] is achieved by sympathetic signals stimulating hepatic glucose production. This balance theory is called the glucostatic theory. In the glucostatic theory the hypothalamus is considered a glucostat and the liver is a unique glucose exchanger, due to the portal system and the hepatic glucose-6-phosphatase. Leptin is dealt with in Chapter 20. Since the hypophysis hormones ACTH and GH are insulin-antagonists the net effect of the hypophysis, when not balanced by a normal pancreatic insulin secretion, is a reduced glucose tolerance. The endocrine pancreas or the pancreatic islets are synonyms for the production site of four polypeptide hormones: Glucagon, insulin, somatostatin, and pancreatic polypeptide (PP). The one million islets of Langerhans are discrete structures scattered throughout the pancreas, but which only comprise 1% of its total weight. The islets are arranged along fenestrated capillaries, so that the hormones can pass easily to the portal blood. The islets of Langerhans receive both sympathetic (adrenergic) and parasympathetic (cholinergic) fibres. The membranes of the islet cells contain gap junctions between neighbour cells, so hormones from one cell can act on its neighbour (paracrine action). Gap junctions allow passage of small molecules from one islet cell to its neighbour. In many pancreatic lobules, the a - b - and d - cells form a paracrine syncytium. 3 a. Glucagon The a-cells of the pancreatic islets is the source of pancreatic glucagon. Glucagon stimulates adenylcyclase in the hepatocytes. This enzyme activates phosphorylase that breaks down glycogen. Actually, glucagon triggers a glycogenolytic cascade, so those considerable amounts of glucose are released in response to the fall in blood glucose. In addition, glucagon stimulates the hepatic production of glucose (gluconeogenesis) from glycerol, alanine and lactate. Glucagon is a direct antagonist to insulin, being catabolic in its actions (gluconeogenetic, glycogenolytic, lipolytic & ketogenic, and deaminating amino acids). Intestinal glucagon (glicentin) is built up from 69 amino acids. The glucagon from the a-cells of the pancreatic islets only contains 29 of the 160 amino acid residues in pro-glucagon. Conditions where there is intracellular lack of glucose (hunger, insulin deficiency, protein rich meals, and amino acid infusion) liberate glucagon from the a-cells of the pancreatic islets to the pancreatic vein and then to the portal vein. Glucagon stimulates ketogenesis (formation of ketone bodies). High blood [glucose] and [FFA] inhibit glucagon secretion. Pancreatic and intestinal (glicentin) glucagon are hepatic insulin-antagonist. Glucagon stimulates hepatic glucose production by glycogenolysis in the hepatocytes and thus increases the blood [glucose]. Glucagon also stimulates gluconeogenesis from glycogenic amino acids in the liver and thus increases urea-genesis. Glucagon stimulates ketogenesis (formation of ketone bodies). In addition to the ketogenic effect, intestinal glucagon is a potent stimulator of insulin secretion - as are other members of the incretin family. Incretins act by increasing cAMP in the b-cells. 3 b. Insulin Banting shared the Nobel Prize with Macleod in 1923 for their work in identifying the role of insulin in the carbohydrate metabolism. Their research led to the practice of insulin therapy for diabetes. Pre-proinsulin is the precursor of insulin. When pre‑proinsulin reaches the endoplasmic reticulum, enzymes separate the molecule from the signal molecule, to form proinsulin. In the Golgi apparatus enzymes cleave proinsulin to insulin (51 amino acids in two chains: A and B) and the C peptide (Connecting peptide). Insulin and C peptide are wrapped in the same secretion granule. The content of these secretion granules is expelled from the cell by exocytosis. When the secretory granules release proinsulin to the portal blood and later the extracellular fluid volume (ECV), connecting peptide (C-peptide) and two amino acids breaks off. The split products are carried to the liver, where half of the insulin molecules are degraded and extracted. The degradation products are broken down and eliminated by the kidney. The kidneys only eliminate c-peptide and its rate of production reflects the rate of insulin secretion. Insulin contains 51 amino acid residues in two chains (m.w. 5734). In healthy persons the blood glucose concentration, B-[glucose], is controlled exactly. The fasting value is within the range of 4-7 mM, with minimum individual variance from day to day, despite varying life conditions with food and exercise. The liver is a glucose exchanger, because it absorbs glucose from the intestine, stores glucose as glycogen, and produce glucose from fat and protein residues (gluconeogenesis). The liver releases glucose to the ECV in exact proportion to the peripheral rate of glucose utilisation in the postabsorptive state (180-200 g per day). The brain metabolism of a healthy standard person requires 100 g of glucose per day. The brain glucose is totally oxidised, ir-regardless of the insulin status. Each meal elicits a peak of insulin secretion, because of the rise in blood [glucose] (Fig. 27-1). The blood [glucose] increases after a meal. Increasing [glucose] is a strong stimulus to the b-cells of the pancreas. Glucose enters these cells through GLUT 2. The cells empty their granules into the ECV, and the granule dissolve immediately after entering the blood. This sequence of events supplies the blood with insulin, C-peptide and proinsulin in the ratio 19:19:1. Insulin reduces the blood glucose for the following reasons: insulin increases the cell uptake of glucose (and potassium) in most tissues (adipocytes, heart and other muscle tissue). The exceptions are the brain, kidney and erythrocytes. The uptake capacity for glucose in hepatocytes is so large, that any insulin effect is immaterial. Insulin promotes the formation of tissue stores from circulating nutrients (the actions are all anabolic). The insulin receptor is a tetrameric protein complex with two a-units extracellularly, and two b-units traversing the membrane of target cells (ie, skeletal muscle fibres, cardiac myocytes and adipocytes). The target cells contain the glucose transporter, GLUT-4. In the absence of insulin, all the GLUT-4 units are located in the intracellular vesicles. Insulin binding to the insulin-receptor activates the tyrosine kinase, which resides in the b-units, and promotes the transport of GLUT-4 vesicles towards the surface of the cell, where they melt together with the membrane. This phenomenon increases the number of glucose-channels through the membrane and thus promotes glucose uptake into target cells. Finally, the insulin-receptor complex is internalised by the cell, insulin is broken down and the insulin-receptor recycles to the cell surface for further use. Glucose is stored in the muscle cells as glycogen, used in the Krebs cycle or it is broken down to lactate. In the fat cells glucose is utilised as a substrate for triglycerides synthesis. In the post-prandial phase, lipolysis liberates fatty acids (FFA) from triglyceride together with glycerol. Glycerol and lactate are substrates for hepatic gluconeogenesis.
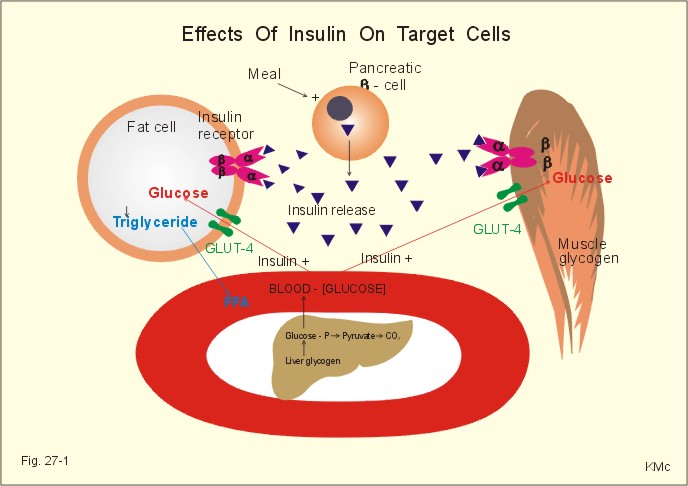
Fig. 27-1: Effects of insulin on target cells. Falling blood [glucose] is called hypoglycaemia, which activates the sympathoadrenergic system and deplete the glucose-dependent brain for its only fuel. Accordingly, the hypoglycaemia causes sympathoadrenergic (sweating, hunger, tremor, tachycardia), and cerebral manifestations (anxiety, disorientation, cramps, and unconsciousness). The clinical picture is that of hypoglycaemic shock. Insulin also increases the rate of glycogen synthesis in the liver and muscles and inhibits the rate of gluconeogenesis. Insulin is a direct antagonist to glucagon being anabolic in its actions (increased glucose entry to cells, increased glycogen and lipid synthesis, decreased protein catabolism and ketogenesis). Glucose-evoked insulin secretion is the result of a chain of events in the pancreatic b-cell (Fig. 27-2).
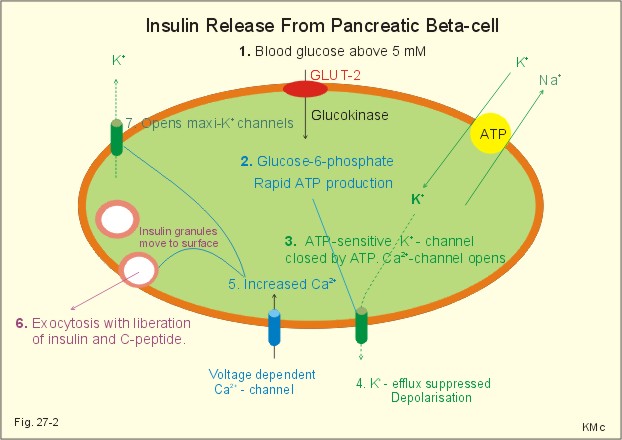
Fig. 27-2: Insulin release from pancreatic b-cell. 1. The glucose uptake takes place through a specific transporter protein (GLUT-2). The pancreatic b-cell membrane contains several K+ channels, of which two are directly involved. This is the K+-ATP channel and the maxi-K+ channel (Fig. 27-2). 2. The hyperglycaemia accelerates the glucose uptake and metabolism and thus increases the ATP/ADP ratio. 3. Increased ATP closes the K+-ATP channels, so the cell depolarises (hypopolarises). During hypopolarisation from the normal resting membrane potential of -70 mV, a threshold is reached at - 50 mV, where the voltage dependent Ca2+ channels open. 4. The Ca2+ influx triggers exocytosis of insulin and C-peptide containing granules following vesicular fusion with the cell membrane. 5. Normally, the maxi-K+ channel and other K+ channels stop depolarisation. When intracellular [Ca2+] and [K+] has increased, it opens the maxi-K+ channel. The K+ efflux restores the resting membrane potential (- 70 mV) towards the equilibrium potential of K+ (-100 mV). Insulin is a vital hormone. Blood from the pancreas passes through the liver, where insulin promotes the production of glycogen from the recently absorbed glucose. The liver destroys a substantial amount of the insulin, whereas the C-peptide passes the liver undisturbed. The plasma [C-peptide] is thus a good estimate of insulin secretion. Insulin can now be synthesized from genetically modified micro-organisms. Insulin is an anabolic hormone. Insulin reduces the blood [glucose] because it increases glycogen synthesis in the liver and muscles. Insulin increases the uptake of glucose through GLUT 4 (in adipocytes, heart and skeletal muscles). Insulin inhibits the gluconeogenesis from glycogenic amino acids in the liver. Insulin promotes the storage of energy, the synthesis of glycogen, mRNA and proteins. Insulin thus reduces urea-genesis. Insulin promotes lipogenesis in the fat stores; however, it inhibits lipolysis. It may be noted that the glycerol portion of the triglyceride molecule is a derivative of glucose. Insulin increases the synthesis of cholesterol in the liver, in particular the rate of VLDL formation (Very Low Density Lipoprotein). The dangerous cholesterol fraction is LDL (Low Density Lipoprotein). Insulin increases the GLUT 4 transfer of glucose and K+ into the muscle cell interior. Certain major tissues (kidney, brain, and intestine) are insensitive to the direct action of insulin. 3 c. Somatostatin D-cells or d-cells are the source of somatostatin, a potent and multipotent hormone inhibitor. Somatostatin contains a disulphide bridge and 14 amino acid molecules. Somatostatin produced in the islets inhibits the local secretion of the other islet hormones, while glucagon stimulates the local release of insulin and somatostatin. Somatostatin is also produced in the hypothalamus, where it functions as the Growth Hormone Inhibiting Hormone (GHIH). Pancreatic somatostatin is released in response to high blood [glucose] and [alanine]. Somatostatin inhibits the secretion of the gastrointestinal tract (but not its motility) and functions as a synaptic transmitter in the CNS. Persons with somatostatin-producing tumours develop diabetes and gallstones. GHIH is synthesized both in the hypothalamic-pituitary system and in the pancreatic islets. The D-cells of the pancreatic islets of Langerhans produce GHIH, which controls the function of the other islet cells by paracrine action. Somatostatin is a multipotent hormone inhibitor. Somatostatin inhibits Somatotropin (GH) but also TSH, insulin, and glucagon. Somatostatin blocks the gastrin secretion in the gastric antrum. Somatostatin inhibits the secretion of digestive fluids, but increases gastrointestinal motility. 3 d. Pancreatic polypeptide The cells responsible for pancreatic polypeptide (PP) secretion are particularly abundant in the head of the pancreas. PP contains 36 amino acid residues in a linear polypeptide. The plasma [PP] increase markedly after a protein rich meal, but it is not released by alanine infusion. The PP secretion is increased by exercise (with high plasma [alanine]), by fasting and by hyperglycaemia. The plasma [PP] is suppressed by glucose infusion. PP inhibits the exocrine pancreas and reduces the gallbladder contractions. This is an appropriate response during exercise and fasting, where any reduction in blood glucose would trigger a PP release. Meals, rich in protein and fat, release pancreatic polypeptides (from the PP-cells). Pancreatic polypeptide inhibits both enzyme secretions from the pancreas and the emptying of bile into the small intestine. This leads to a delay in the absorption of nutrients including glucose. Patients with pancreatic islet cell neoplasm have elevated plasma [PP]. 4. Pancreatic exocrine control Endocrine glandular tissue is localised in the Langerhans islets that produces insulin in the b-cells, glucagon in the a-cells, somatostatin (GHIH) plus gastrin from the d- and G-cells, and pancreatic polypeptide (PP) from the P-cells (Fig. 27-3). Bombesin, galanin, and neuropeptides are present in pancreatic neurons and act as transmitters. Stimulation of vagal fibres to the pancreas enhances the rate of enzyme secretion into the pancreatic juice. Stimulation of sympathetic fibres reduces bloodflow to the pancreas, and thus inhibits pancreatic secretion. Gastrin enhances enzyme secretion and insulin potentiates the effect, whereas somatostatin inhibits secretion from both acinar and ducts cells (Fig. 27-3: - all).
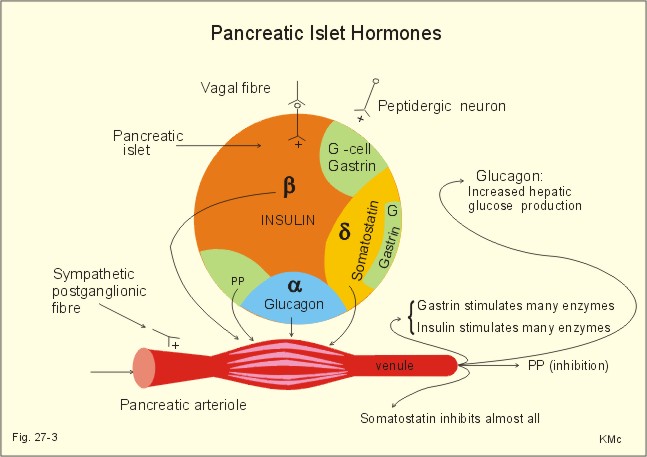
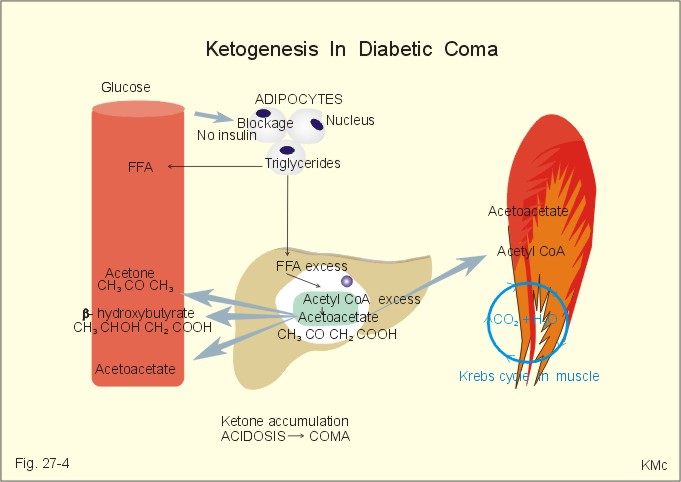
Fig. 27-3: Liberation of pancreatic islet hormones. Diabetes mellitus (DM) DM is a collective term for a multitude of metabolic disorders, where lack of insulin (type I diabetes) or insulin resistance (type II) dominates. Insulin resistance is defined as insufficient sensitivity to insulin. The diabetic condition is characterized by an abnormal glucose tolerance that is documented by the use of a glucose tolerance test. Besides hyperglycaemia, the overall phenomena in the diabetic condition are protein depletion and increased lipolysis with deposition of lipids in the vascular walls of the brain, heart, kidneys, eyes and muscles. All cases of DM where the cause is not fully explained are termed primary DM, whereas secondary DM is explainable and sometimes directly curable. Secondary DM is caused by hypersecretion of one or more of the many catabolic hormones with hyperglycaemic effect (adrenaline, noradrenaline, glucagon, glucocorticoids and growth-hormone, HG) or by total destruction of the pancreas from pancreatitis or carcinoma. The hormone disorders are phaeochromocytoma, glucagonoma, Cushing’s syndrome and acromegaly. Most of the patients with primary DM can be classified into the two groups already presented above Insulin-Dependent DM (IDDM) or type I diabetes, and Non-Insulin-Dependent DM (NIDDM) or type II diabetes. This paragraph deals with 1. IDDM, 2. NIDDM, 3. Insulin shock (hypoglycaemia), 4. Oral and intrveneous glucose tolerance tests, 5. treatment of diabetes mellitus, and 6. Summary of the diabetic condition. The presentation of IDDM is typically a young person with a few week history. This is a serious, life threatening metabolic disease, where continuation of life depends upon insulin treatment. The first treatment with insulin took place in Canada (1922). Until then these patients died within half a year in ketoacidotic coma. Persons, often with hereditary predisposition, are suddenly attacked by autoimmune destruction of all b-cells in the pancreatic islets, which results in the complete absence of insulin. This autoimmune destruction occurs more often in populations, where breast feeding is unpopular, and protein-rich cow's milk is used generally. Lack of insulin leads to extracellular hyperglycaemia, and increased lipolysis. The classical triad is: 1. Polyuria (osmotic diuresis due to extracellular hyperglycaemia and glucosuria). 2. Polydipsia and thirst due to the loss of salt and water. 3. Weightless due to extracellular fluid volume (ECV) depletion and the breakdown of tissue stores (ie catabolic effect of insulin deficiency) with rapid wasting. The intracellular lack of glucose activates glycogenolysis in the liver and muscle cells; lipolysis and muscular proteolysis is accelerated. The liberated free fatty acids (FFA) are converted to ketone bodies, whereby a metabolic acidosis (ketoacidosis) is produced. A patient with a blood (glucose) above 25 mM loses consciousness and to such degrees that contact is impossible and reactions to pain are absent (coma). Diabetic ketoacidosis is a condition of insulin deficiency causing increased hepatic ketogenesis. This condition of uncontrolled catabolism occurs in IDDM, only. In a young person, the first sign of IDDM can be coma with diabetic ketoacidosis - a life-threatening condition, if the patient is alone. This is particularly likely, when the patient is under the stress of intercurrent illness such as infection with high fever. Also a recognized IDDM patient can be hit by ketoacidosis during intercurrent illness, where his insulin demand is increased, or the patient may take too little insulin because of lost appetite or for any other reason. Insulin deficiency has two consequences. First of all, the hepatic glucose release accelerates, and secondly the uptake of glucose by muscle and fat cells in the periphery is reduced. Progressive hyperglycaemia causes osmotic diuresis with loss of salt and water. The abnormally low ECV is termed the dehydrate state, and with falling blood volume also renal bloodflow falls. Insulin deficiency also accelerates the lipolysis (Fig. 27-4).
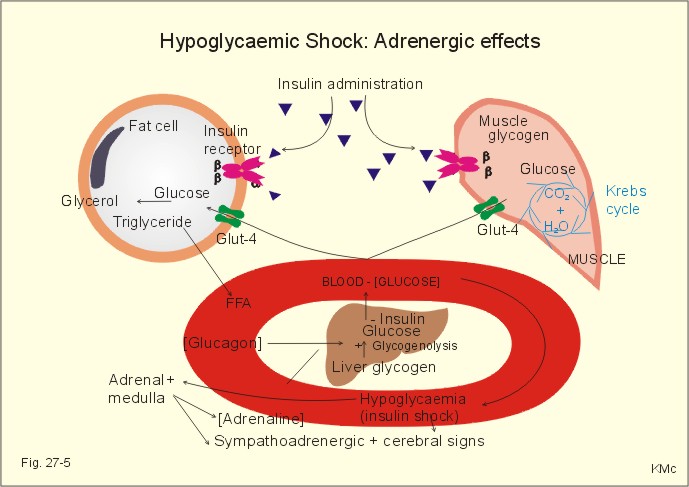
Fig. 27-4: The development of ketoacidosis during insulin deficiency (FFA = free fatty acids). Triglycerides are liberated from adipose tissue, and the concentration of free fatty acids (FFA) in the blood is elevated. FFA are broken down to fatty acyl carnitine within the liver cells, and this molecule is converted to acetyl CoA, which in turn reach the mitochondria, where ketone bodies (aceto-acetate, acetone, b-hydroxybutyrate) are formed (Fig. 27-4). The breath of the patient smell by acetone, and there is ketosis in the urine. The concentration of ketone bodies in the blood passes 5 mM, and when pH falls below 7, there is life-threatening or terminal coma. The condition is called an acute metabolic acidosis, characterized by a negative base excess. The patient tries to compensate the metabolic acidosis by hyperventilation, so-called Kussmaull-breathing. Most patients with recent IDDM have circulating antibodies to islet cells, and tend to develop other organ-specific autoimmune disorders (Addison disease, Hashimotos thyroiditis and pernicious anaemia). Identical twins show a 40% concordance in developing IDDM, so life-style must also play a role. There is an association with HLA-DR4, if also HLA-B8 or HLA-DR3 is present. This is a frequent type of DM in particular in populations with a sedentary life style and obesity. The incidence increases with age and development of obesity, which is reflected in the name maturity-onset diabetes. The prevalence is high in Afro-Caribbean and Asian population groups. The onset of NIDDM is sometimes triggered by Intercurrent illness or by pregnancy, but not by immunological reactions. NIDDM is a complex of polygenic disorders. Certain families show an autosomal dominance. the genetic defects differ and many mutations are known. One is the gene on chromosome 7, which code for glucokinase. Identical twins show almost absolute concordance in development of NIDDM. The much more frequent type II diabetes (maturity-onset) is the result of insulin resistance and b-cell defects. Type II diabetes also occurs in younger persons, especially in persons with a high fat-low muscle mass. A strong genetic element is always present, but inactivity and stress (an inactive life style with a low endurance capacity) seems to be involved in the development of type II diabetes. - Lack of exercise predisposes one to obesity, a condition that greatly decreases insulin sensitivity of the target cells (adipocytes, heart and skeletal muscle tissues). Reduced glucose combustion creates hyperglycaemia. The hyperglycaemia elicits insulin secretion from defective b-cells in some patients, resulting in raised serum [insulin]. Since insulin is present, the acute complications such as ketonaemia and metabolic acidaemia (often found with IDDM) are rare in these patients. The high serum [insulin] may further down-regulate the activity of their insulin receptors. The insulin secreted in NIDDM patients does not increase the uptake of glucose as in normal persons. Many NIDDM patients need much more insulin for a given test effects than IDDM patients and healthy people. An inactive life style for years, with redundant food intake, seems to be involved in the development of NIDDM in persons with a genetic predisposition. Lack of regular physical activity with development of overweight, increases the incidence if NIDDM. The impaired glucose tolerance is demonstrated by a glucose tolerance test. The insulin secretion is abnormal in patients with NIDDM, although they typically possess half of their b-cell mass at autopsy. The destroyed b-cells is filled with amyloid material (islet amyloid polypeptide, IAPP). IAPP is a possible antagonist to insulin, and explain some cases of insulin resistance. Many older patients with NIDDM have no symptoms, but a routine examination reveals glucosuria or a raised blood [glucose]. Other patients are tired, have minor genital infections or sugar spots on their underwear. Some patients present with established late-complications such as retinopathy (blindness), nephropathy, arteriosclerotic disorders (cerebrovascular insults, myocardial infarction, intermittent claudication, gangrene), susceptibility to infection or neuropathy. NIDDM can be caused, theoretically, by 1. b-cell defects including genetic defects, resulting in abnormal insulin production, or by 2. target cell defects including receptor failure. The possible defective sites in 1. and 2. have one common denominator. They are all key proteins (hormone, receptors and transporters). Muscular activity is required to stimulate the normal production of key proteins. NIDDM relates to inactive life style. The basic problem is therefore possibly a genetically and activity dependent defect in key protein production in the cell interior. Actually, a genetic defect has just been demonstrated at certain steps of insulin action in a subset of patients of late-onset NIDDM. 3. Insulin shock (hypoglycaemia) A high blood [insulin] will cause tissues to store away the available blood glucose rapidly, mainly through muscular GLUT 4, and stop simultaneously the production of new glucose. A low blood glucose level elicits a large secretion of glucagon to the portal blood. Glucagon is the most important insulin-antagonist. Glucagon increases hepatic glucose production (enhancing glycogenolysis, gluconeogenesis and protein breakdown). Low glucose levels trigger glucagon production, even from denervated, pancreatic a- cells; hence, they must be glucose sensitive. An increased catecholamine secretion from sympathetic nerve endings (NA) and Ad from the adrenal medulla (elicited from the hypothalamic glucostat via the sympathetic nervous system) helps within minutes to compensate, as catecholamines stimulate glycogenolysis, increase lipolysis and inhibit peripheral glucose uptake. Hours later, cortisol and GH also contribute. An appropriate rise of plasma [cortisol] in response to insulin-induced hyperglycaemia documents an intact CRH-ACTH-adrenal axis, and this is the most widely used stress test. Adrenergic effects, such as trembling fingers, tachycardia, and muscular stiffness warn the hypoglycaemic patient. The glucose consumption by the heart and brain continues. The lack of glucose in the brain makes the patient uneasy at first; he is then insecure, anxious and has cold sweat. Later in hypoglycaemia, the patient becomes confused, furious and denies with slurred speech to take glucose. Blood [glucose] below 2.5 mM elicits hypoglycaemic shock with loss of consciousness (somnolence, sopor or coma), universal cramps and respiratory stop (Fig. 27-5). Intravenous injection of glucose (50%) is the rational therapy for hypoglycaemic coma. The patients wake up almost immediately, and are then often in a hyperglycaemic state.
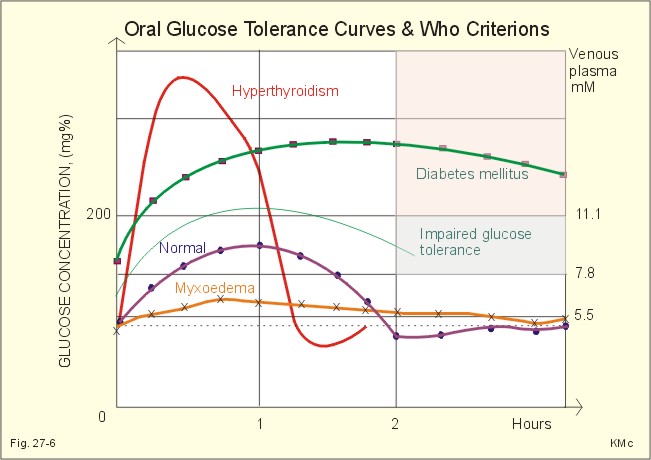
Fig. 27-5: Consequences of hypoglycaemic shock. The b-cell defects are insufficiently described. Type 2 diabetics do not produce sufficient levels of ATP in the pancreatic b-cells to completely block the K+ -ATP channels (Fig. 27-2). Thus, the b-cell does not hypopolarize adequately in response to hyperglycaemia. Therefore, the voltage dependent Ca2+ -channels is insufficiently activated, and intracellular [Ca2+] do not increase enough to trigger the insulin exocytosis needed. Sulfonylurea compounds close the K+-ATP-channels and thus help to treat type 2 diabetes. 4. Oral and intravenous glucose tolerance tests The test is performed orally or intravenously. Oral test. The patient drinks a glucose solution containing 75 g glucose within four minutes (WHO). The blood concentration in venous plasma ([glucose]) is followed over 3 hours by blood sampling. Normal individuals start from a low fasting [glucose] such as 5.5 mM or less. The blood [glucose] peaks after one hour and returns to normal within two hours (less than 6.7 mM in Fig. 27-5). Persons with impaired glucose tolerance start with a fasting level less than 7.8 mM, and after 2 hours the level is 7.8-11.1 mM.
Fig. 27-5: Oral glucose tolerance curves for a normal person, a diabetic, a patient with hyperthyroidism and a patient with myxoedema. The diabetic patient typically starts from a high fasting [glucose], such as values above 7.8 mM, and increase to a very high level. The blood [glucose] is not back to normal within two hours, but stays above 11.1 mM. This test pattern is the clinical WHO criterion of diabetes (Fig. 27-5). A patient with hyperthyroidism (Graves disease or Morbus Basedowii) has a rapid intestinal absorption and a rapid combustion of glucose. The myxoedematous patient has a slow absorption and a slow combustion of glucose.
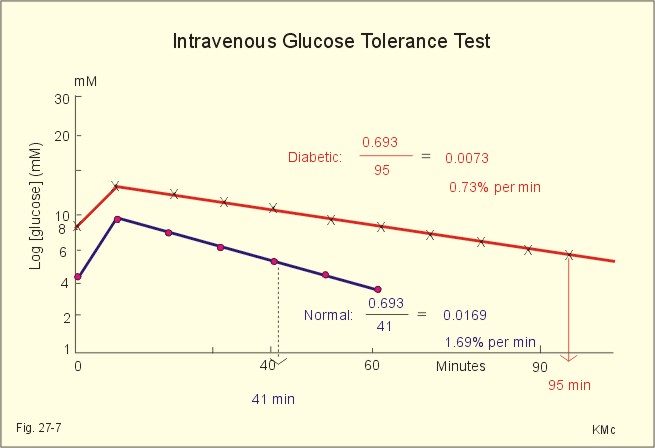
Fig. 27-6: Results of i.v. glucose tolerance tests from a normal person and from a diabetic. Intravenous (i.v.) test. We inject 25 g glucose intravenously over a period of 4 minutes. We then measure the blood [glucose] every 10 min for at least an hour in order to determine the half-life from a semi-logarithmic plot (Fig. 27-6). The metabolic combustion rate for glucose is exponential, so it is easy to calculate the metabolic rate constant (k) expressed in percentages. Note that the metabolic rate constant k here is the amount of glucose combusted divided by the total amount of glucose in a mainly extracellular distribution volume. The half-life (T1/2) is equal to 0.693/k. All glucose combustion rates above 1.2% per min are normal.
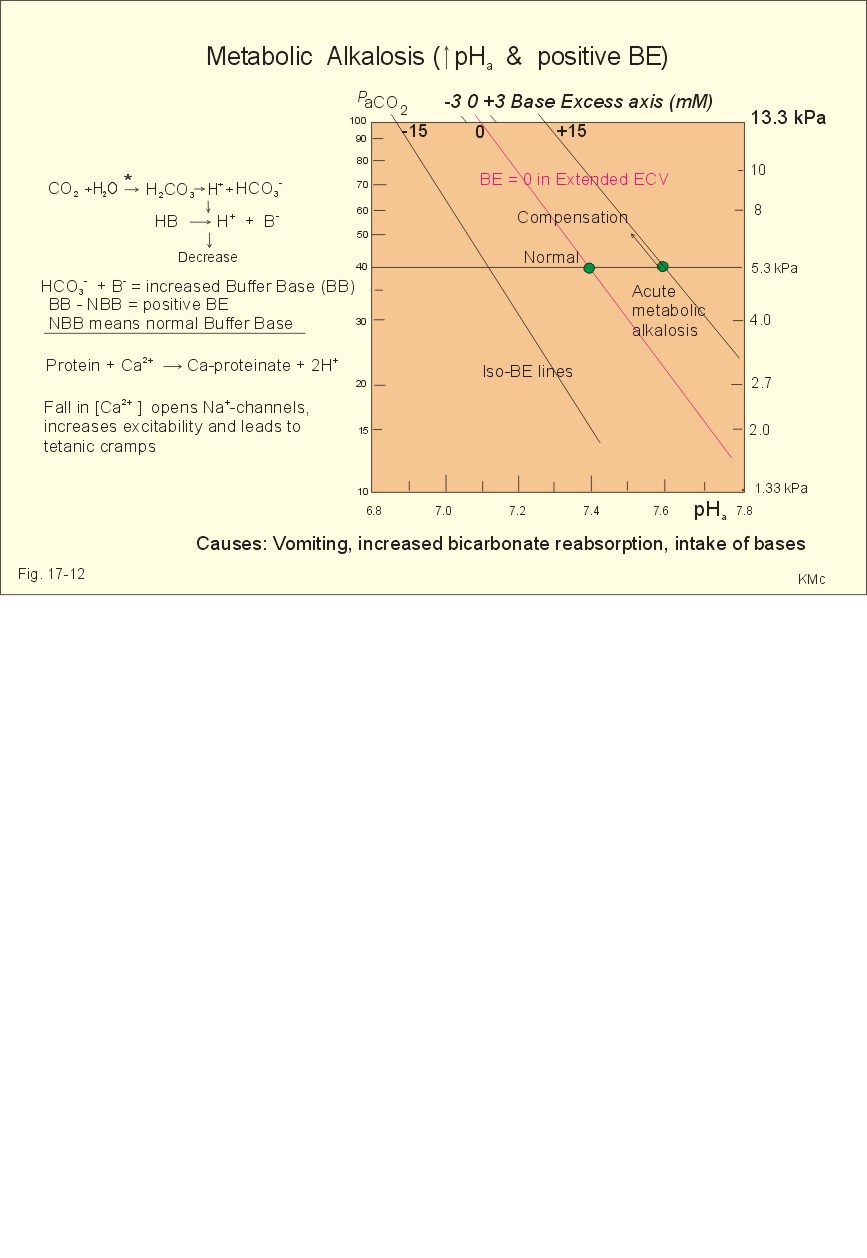
Fig. 27-7: Intravenous Glucose Tolerance Test 5. Treatment of diabetes mellitus A normal person with three meals per day will have three peak concentrations of glucose and insulin in his blood. It is possible to obtain such a time profile in a diabetic person by the following strategy. Inject a fast‑acting insulin three times a day just before meals and a slow‑acting insulin at night. This is the physiologic principle. The aim of this procedure is to reduce the number of acute and chronic complications for diabetics. All diabetics are recommended to eat healthy just like anyone else. When diet alone is insufficient to achieve a satisfactory blood glucose, a slim type II diabetic is treated with sulphonylurea compounds. The obese type II diabetic is treated with a biguanide called metformin. Patients, who have been in metabolic acidosis, are usually treated best with insulin. 6. Summary of the diabetic condition A poorly controlled diabetic condition leads to extracellular hyperglycaemia, glucosuria, metabolic acidosis, polyuria (osmotic diuresis), dehydration and polydipsia. The osmotic diuresis leads to the excretion of Na+ and water, which results in Na+ and ECV depletion. Intracellular lack of glucose activates glycogenolysis in the liver and muscles, and accelerates muscular proteolysis and lipolysis. This liberates free fatty acids, which are converted to ketone bodies. A patient with hyperglycaemia above 25 mM loses consciousness to such a degree that contact is impossible (ie, coma). The increased rate of cholesterol production increases the occurrence of atherosclerosis and of diabetic nephropathy. Albuminuria, hypertension and low GFR characterise diabetic nephropathy. I. Each of the following five statements have True/False options: A. Peptides and protein hormones are lipophobic. B. Infertility is a diagnosis used on a couple, which have been unable to conceive during one year of unprotected intercourse. C. Chronic hypoadrenalism is also called Addison’s disease. D. Nephrogenic diabetes insipidus, is a condition where the renal cells are resistant to ADH. E. Glycerol and lactate are substrates for hepatic gluconeogenesis. II. Each of the following five statements have True/False options: A. Receptors are frequently glycosylated, so one signal molecule linked to a receptor is always enough to elicit a response. B. The incidence of atherosclerosis is correlated to the total [cholesterol], the [LDL], and the [LDL]/ [HDL] ratio in blood plasma. C. Oestrogens, exercise, nicotinic acid and alcohol increase the plasma-HDL. D. Omeprazole stimulates the luminal gastric proton-pump. E. Prostaglandins are primarily paracrine hormones, which act through receptors linked to G-proteins. A 19 year old male, body weight 80 kg, was suddenly complaining of fever during his work and ordered home to bed. The patient was living alone. Fortunately a colleague visited him the next morning. He had to break the door down and found the patient unconscious. The patient arrived at the hospital in deep coma. A blood sample from the radial artery showed the following results: b-cell antibodies, PaCO2 27 mmHg, pH 7.21, actual bicarbonate 10.5 mM, O2 saturation 0.96 and [glucose] 32 mM (5.75 g/L). The Base Excess is -15 mM in the extracellular fluid volume (see Fig. 17-12). The urine contained glucose and ketone bodies. The patient's breathing was deep and fast, his heart rate was 115 beats/min, and his blood pressure was 90/55 mmHg. The mucous membranes of the mouth were dry and the tonsils were enlarged and infected. The rectal temperature was 39.9 Centigrade. 1. Explain the condition of the patient concerning thermo-balance, carbohydrate metabolism, acid-base balance and fluid balance. 2. Describe a rational treatment of the four homeostatic disturbances mentioned in 1. 3. Following eight hours of treatment the blood pH was 7.41 and PaCO2 42 mmHg, but the patient is still hyperventilating. Explain why. 4. Following 24 hours of treatment all blood gas values were normal and the patient was resituated. Why did the patient stop to hyperventilate? A female of 49 years and with a height of 1.52 m is in hospital due to obesity and related problems. Her weight is 74 kg. She has developed skin mycoses and multiple boils. In the morning (fasting state) a blood sample shows a blood [glucose] of 7.4 mM, but glucose is not found in the urine. At the hospital her total body water is measured following intravenous injection of radioactive water (5.5 × 107 Becquerel or Bq tritium water). Her bladder is emptied at the time of injection. Two hours later the bladder is drained for 95 ml of urine with a concentration of 1 598 400 Bq per l. At this time the indicator is evenly distributed in the total body water with a concentration of 1 520 700 Bq per l. The amount of indicator lost in the urine is 2/3 of the total loss. 1. What is the principle for estimation of total body water? 2. Calculate the total body water in litres and in fraction of her body weight. 3. Is this a normal result? 4. The patient is obese, but is this a serious overweight? 5. Does she have symptoms and signs of complications? A 23-year old male was saved after 30 days in the ruins of a house following earth quake. There was no food but sufficient water. At the arrival to the hospital the patient was in syncope with frequent, deep respiration, and the expired air smelled of acetone. The skin was dirty with brown pigmentation. The cardiac rate was 85 bpm, and the arterial blood pressure was 11.3/7.3 kPa (85/55 mmHg). The blood [glucose] was 2.2 mM, and the plasma [FFA] was increased. The serum concentrations of proteins and essential amino acids were reduced. The blood [haemoglobin] was 95 g l-1. There was moderate antidiuresis with ketonuria with signs of water retention and a high nitrogen loss in the urine. The patient was treated with parenteral administration of glucose, amino acids and electrolytes. Following the glucose intake, the blood [glucose] was increased to 10 mM, and glucosuria occurred. A glucose tolerance test was performed and resulted in a high blood [glucose] level that had not reached the normal level within 2 hours.
See answers · Glucose is absorbed through the luminal membrane of the intestinal cells in glucose-Na+ transporter proteins. The two substances pass through the basolateral membrane via separate routes: Glucose passes in a special glucose-transporter, and Na+ is transferred by the Na+-K+‑pump. · Somatotropin ‑ human growth hormone (GH) ‑ is an insulin-antagonist, but together with insulin probably the most important anabolic hormone. · Glucose sensitive neurons in the hypothalamus (the glucostatic centre) react to hypoglycaemia by releasing glucagon from the pancreatic a-cells and catecholamines from the adrenal medulla by action of the sympathetic system. · Since the hypophysis hormones ACTH and GH are insulin-antagonists the net effect of the hypophysis, when not balanced by a normal pancreatic insulin secretion, is a reduced glucose tolerance. · The endocrine pancreas or the pancreatic islets are synonyms for the production site of four polypeptide hormones: Glucagon, insulin, somatostatin, and pancreatic polypeptide (PP). · Insulin is synthesized as proinsulin, which is stored in granules close to the cell membrane of the b-cells of the pancreatic islets. When the secretory granules release proinsulin to the portal blood and later the extracellular fluid volume, connecting peptide (C-peptide) and two amino acids breaks off. · A poorly controlled diabetic condition leads to extracellular hyperglycaemia, glucosuria, metabolic acidosis, polyuria (osmotic diuresis), dehydration and polydipsia. The osmotic diuresis leads to the excretion of Na+ and water, which results in Na+ and ECV depletion. · Intracellular lack of glucose activates glycogenolysis in the liver and muscles, and accelerates muscular proteolysis and lipolysis. This liberates free fatty acids, which are converted to ketone bodies. · A patient with hyperglycaemia above 25 mM loses consciousness to such a degree that contact is impossible (ie, coma). · The increased rate of cholesterol production increases the occurrence of atherosclerosis and of diabetic nephropathy. · Albuminuria, hypertension and low glomerular filtration rate characterise diabetic nephropathy. Almind, K., C. Bjørbæk, H. Vestergaard, T. Hansen, S. Echwald, and O. Pedersen. "Aminoacid polymorphisms of insulin receptor substrate-1 in non-insulin-dependent diabetes mellitus." The Lancet 342: 828-832, 1993. Ashcroft, F.M. and S.J.H. Ashcroft. "Insulin." IRL Press at Oxford Univ. Press, Oxford 1992. Banting, F.G. and C. H. Best. "Internal secretion of pancreas." J. Lab. and Clin. Med. 7: 251-326, 1922. Flatt, P.R. "Nutrient regulation of insulin secretion." Portland Press Ltd., London 1991.
|
||
.jpg){kind=link}
{kind=link}
Click here to introduce your comments or contributions