New Human Physiology | Paulev-Zubieta 2nd Edition
Chapter 31: The Human Genome and Genetic Disorders
| HOME | PREFACE | TABLE OF CONTENTS | SYMBOLS | SECTION INFO | CONTRIBUTORS | LINKS | CONTACT US |
Highlights
Study_ObjectivesPrinciplesDefinitionsEssentials
PathophysiologyEquationsSelf-AssessmentAnswers
Further Reading
|
Chapter 31
|
 |
|
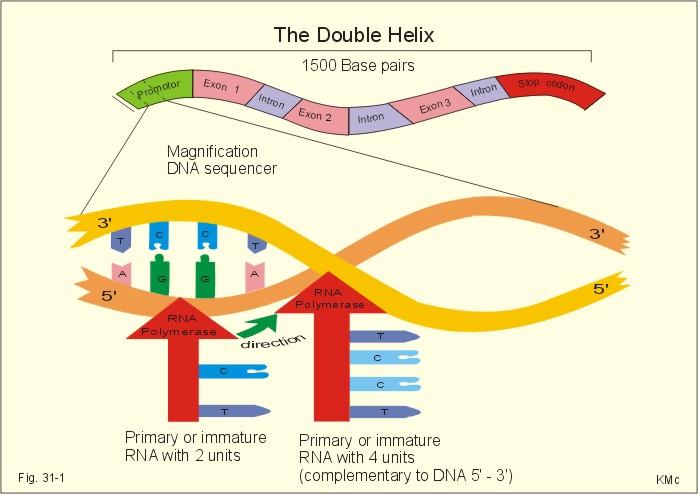
The Human Genome And Genetic Disorders This Chapter was written following fruitful discussions with my colleague, Erik Niebuhr, MD, DSc, University of Copenhagen. · To define allele, anaphase, anticodon, autosomes, chromatid, chromatin, chromosome, clone, codon, diploid, exons, gene, gene frequency, haploid, haemophilia, introns, nucleotides, post-translational modification, probability, promoter sequence, ribosomal translation, sex-linked genes, splicing, transcription, translocation, and transversion. · To describe the human genome, gene therapy, and the roles of DNA, RNA, messenger RNA, transfer RNA and recombinant DNA · To calculate the frequency of abnormal genes in the total gene pool of a population. · To explain protein synthesis, DNA transcription, mutations, and genetic disorders. · To use the above concepts in problem solving and case histories. · Many hormones regulate genes expressed by some cells. This mechanism controls the synthesis of enzymes, receptor proteins, structural proteins, and transcription proteins. This is why steroid and thyroid hormones require hours for their biological effect. · The partial or total lack of specificity in the third base of some triplet codons may allow 2-4 codons, differing only in the third base, to code for the same amino acid. · Allele is an alternative form of a gene occupying the same locus on a particular chromosome. · Anaphase refers to the stage of nuclear division, which is characterized by movement of chromosomes from spindle equator to spindle pools. · Anticodon is the group of three nucleotides in transfer RNA that pairs complementarily with three nucleotides of messenger RNA during protein synthesis. · Autosomes refers to any chromosome which is not a sex chromosome or mitochondrial chromosome. Humans possess 22 pairs of autosomes. · Bacteriophag is a bacterial virus. Bacterial viruses are modified and used as vectors for DNA cloning. · Chromatid results from the replication of chromosomes during interphase. A chromatid is one of the two identical halves of a chromosome, which shares a common centromere with a sister chromatid. · Chromatin refers to the nuclear material, which comprises the chromosomes: The DNA complex. · Chromosomes are nucleoprotein structures, which are the sites of nuclear genes arranged in linear order. · Clone. A group of cells or organisms derived from a single ancestral cell or individual by asexual multiplication (repeated mitoses). All members of a clone are genetically identical. · Codon or triplet code is determined by a base triple (three adjacent nucleotides that code for one amino acid). A codon encodes a specific amino acid residue to be added into the peptide chain or specifies termination of translation. Three codons in mRNA (UAA, UAG and UGA) are stop codons. The signals encoded by the codons form the genetic code. · Dalton refers to a weight unit equal to the mass of the hydrogen atom. · Deoxyribonucleic acid (DNA) is a helical coiled nucleic acid molecule composed of deoxyribose-phosphate units connected by paired bases attached to the deoxyribose sugar. DNA is the genetic material of all living organisms and vira. · Diploid refers to the number of chromosomes found in somatic cells (ie, two sets). · DNA polymerase is the enzyme that replicates DNA. · Dominant describes a trait expressed in individuals who are heterozygous for a particular gene · Exon is a segment of a gene that is represented in the final spliced mRNA product. · Expressivity is the degree to which the effect of a gene is expressed. · Frequency refers to the relative number of actual cases per 100 000 persons in a population. · Gene is a part of a DNA molecule that codes for the synthesis of a specific polypeptide chain through its sequence of nucleotides. Each human gene extends over 40 kb in general, but we also posses longer genes. The gene, located in the chromosome, is the particulate determiner of hereditary trait. · Gene frequency refers to the number of loci at which a gene occurs, divided by the number of loci at which it could occur. This is the proportion of one allele of a pair of genes present in the population. · Gene therapy is the application of gene transfer (DNA delivery to cells) in an attempt to treat genetic or acquired disorders. The transferred DNA must contain promoter zones for transcription and the protein-coding region of the gene. · Genome is the total amount of genetic material in the cell. · Genotype is the genetic constitution of an individual. · Haploid refers to an individual or germ cell having a single complete set of chromosomes (one set). · Heterozygote is an individual possessing two different alleles at the corresponding loci on a pair of homologous chromosomes. · Homozygote is an individual possessing identical alleles at the corresponding loci on a pair of homologous chromosomes. · Haemophilia is an X-linked, recessive genetic disorder characterised by free bleeding from even slight wounds because of lack of formation of clotting substance. · Incidence refers to the new cases of a disorder diagnosed per year in a population group. · Intron is a segment of a gene not represented in the final mRNA product. The segment has been removed through splicing together of exons on either side of it. · Karyotype is the number, size and shape of the chromosomes in a cell. · Meiosis is a nuclear division in which the diploid chromosome number is reduced by half. · Mutagens mean all agents that bring about a mutation. · Mutation is a sudden change in genotype without relation to the ancestry of the individual. · Nucleotides are the basic units of nucleic acids and contain a 2-deoxyribose (a pentose sugar molecule), a phosphate group, and a nitrogenous base. DNA has only four types of nitrogenous base, namely: Adenine (A), Cytosine (C), Guanine (G), and Thymine (T): A C G T. · Oncogenes are genes which are altered in structure or expression, and contribute to the neoplastic transformation of cells (cancer cells). · Phenotype is the appearance of an individual, resulting from the effects of both environment and genes. · Post-translational modification: Following ribosomal translation, proteins are modified by addition of carbohydrates, cleavage of bonds within the new protein, shortening or folding. · Polyploid refers to an individual having more than two complete sets of chromosomes (triploid = 3, tetraploid = 4). · Probability is the likelihood of occurrence of a given event. · Promoter is a region of DNA that plays an essential role in the initiation of transcription of a gene. · Recessive describes a trait expressed in individuals, who are homozygous for a particular gene but not found in the heterozygote. · Recombinant DNA refers to a DNA strand resulting from the physical joining of two or more pre-existing DNA strands. · Ribonucleic acid (RNA) is similar to DNA except that it contains ribose instead of pentose and Uracil (U) instead of T: A C G U. RNA is usually single stranded. · Ribosomal translation is programmed protein synthesis. Inside the ribosome the particular sequence of the mRNA is read, and the particular sequence of nucleotides is build into a polypeptide. This is the so-called translation process, whereby the amino acids are always linked to the polypeptide chain in the proper sequence. · RNA polymerase is the enzyme that synthesises RNA based on a DNA template. · Sex-linked gene is a gene located on the X- or Y-chromosome in XY species. The genes of well-known sex-linked disorders are located mainly on the X-chromosome. · Splicing takes place inside the cell nucleus. The coding sequences or exons of the immature RNA molecule are cut and spliced together by enzymes eliminating intervening introns. The final product is called mature messenger RNA (mRNA). · Transcription (copying) is the copying of the DNA code into a single stranded immature messenger RNA (mRNA). RNA is ribonucleic acid. In the TATA box, the RNA polymerase transcribes a single stranded copy of the DNA sequence. The copying stops at the end of the gene. · Transfer RNA (tRNA) is a molecule which carries a single amino acid and transfer the amino acid to the ribosome. - There are at least 20 different types of tRNA, each carrying only one amino acid. · Translation is the process by which a specific mRNA nucleotide sequence is responsible for a specific amino acid sequence of a polypeptide. The genetic information is translated into protein synthesis. · Translocation refers to the transfer of a portion of one chromosome to another non-homologous chromosome. · Transversion is the substitution in DNA or RNA of a purine for a pyrimidine or vice versa. · Trisomy is the representation of a chromosome three times rather than twice, yielding a total of 47 chromosomes. · Vector is a DNA molecule used to carry DNA regions. · Zygote is the result of fusion of two gametes in sexual reproduction (a fertilised egg is a zygote). This paragraph deals with 1. The human genome, 2. Protein synthesis, 3. DNA transcription. Genetic coding is stored in DNA or deoxyribonucleic acid (Fig. 31-1). Nuclear DNA is the dominant controller of protein synthesis in the ribosomes. The codes of special enzymes and other proteins are based on at least 20 amino acids arranged in sequence. The nucleus contains a nucleolus and the chromatin network. Chromatin is DNA molecules rolled up in a pearl chain structure with proteins called nucleosomes. DNA consists of two nucleotide strands twisted around each other to form the so-called double helix (Fig.31-1), which can be several cm in length. The double helix is folded to form chromosomes with a length of approximately 10 mm (Fig. 31-1). Nucleotides contain a 2-deoxyribose (a pentose sugar molecule), a phosphate group, and a nitrogenous base. DNA has only four types of nitrogenous base, namely: Adenine (A), Cytosine (C), Guanine (G), and Thymine (T): A C G T (Fig. 31-1) The human body cell contains 46 chromosomes in a normal nucleus. Females have two X-chromosomes and two sets of 22 autosomes, whereas males have one X- and one Y-chromosome plus two sets of 22 autosomes. The length of a DNA molecule is measured as the number of base pairs (bp), and a fraction of DNA which is 1000 bp long is called one kb (1 kilobase pair). A gene is a part of a DNA molecule that codes for the synthesis of a specific polypeptide chain through its sequence of nucleotides. Each human gene extends over 40 kb in general, but we also posses longer genes. A portion of a human gene - showing 1.5 kb - is drawn in Fig. 31-1.
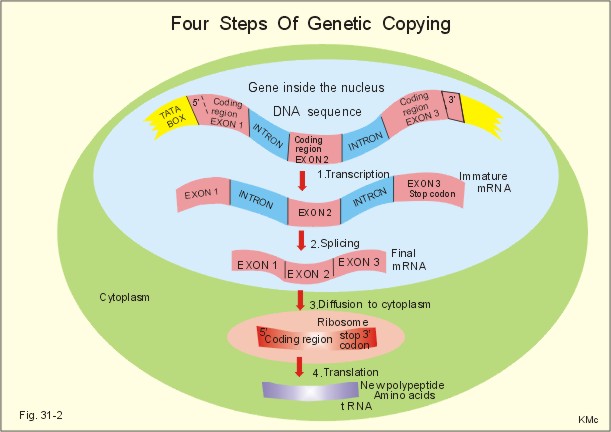
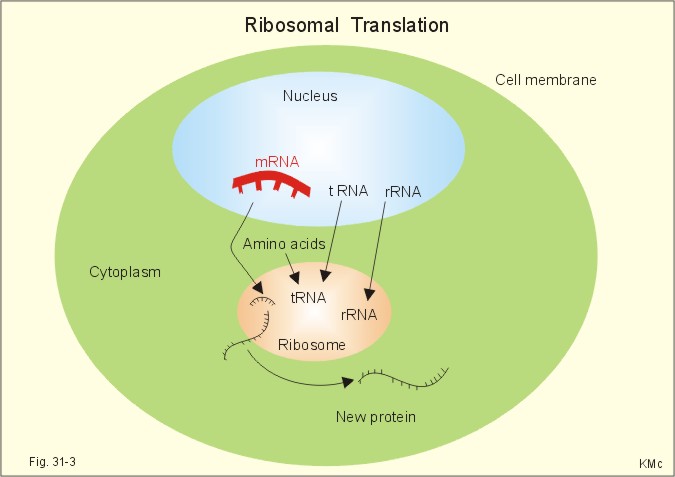
Fig. 31-1: DNA sequence with nitrogenous bases inside the double helix. The complementary base pairs are held together by weak hydrogen bonds. The drawing is 2-dimensional, whereas reality is 3-dimensional. Thus, the distance between the two DNA strands is approximately identical at any point along the DNA spiral. The details are magnified from the promoter zone of the gene. The entire human genome on the 24 different chromosomes is estimated to be 3*109 bp long and to contain 105 genes. Each gene has its own promoter sequence. A promoter sequence is a particular sequence of A, C, G, and T (Fig. 31-1). The nuclear enzyme RNA polymerase recognises a particular sequence in the promoter region, and binds to a region of TATAAT, also called the TATA box of the promoter region. 2.1. Transcription (copying). This is the copying of the DNA code into a single stranded immature messenger RNA (mRNA). RNA is ribonucleic acid. In the TATA box, the RNA polymerase transcribes a single stranded copy of the DNA sequence. The transcription process (copying) stops at the end of the gene. RNA is similar to DNA except that it contains ribose instead of pentose and Uracil (U) instead of T: A C G U. RNA is usually single stranded. The RNA polymerase activity is shown in two stages, first with 2 base units and later with 4 (Fig. 31-1). 2.2. Splicing. The so-called splicing also takes place inside the cell nucleus (Fig. 31-2). The coding sequences or exons of the immature RNA molecule are cut and spliced together by enzymes eliminating intervening introns (ie, non-coding sequences). The final product is called mature messenger RNA (mRNA). 2.3. Diffusion. The mRNA diffuses through the nuclear membrane into the cytoplasm, where it is bound to ribosomes (Fig. 31-2). 2.4. Ribosomal translation is programmed protein synthesis. Inside the ribosome the particular sequence of the mRNA is read, and the particular sequence of nucleotides is build into a polypeptide (Fig. 31-3). This is the so-called translation process, whereby the amino acids are always linked to the polypeptide chain in the proper sequence (Fig. 31-2). Following ribosomal translation, proteins are modified by addition of carbohydrates, cleavage of bonds within the new protein, shortening or folding. These processes are called post-translational modification (Fig. 31-3).
Fig. 31-2: Four steps of genetic copying from DNA in the nucleus to new protein in the cytoplasm via mRNA. The first nucleotides on the mRNA form a regulatory sequence, and do not code for amino acids. This sequence is also called the 5'untranslated region (Fig. 31-2, green 5'). The nucleotide sequence in the middle of the mRNA code for amino acids - the so-called coding region. The end of the mRNA does not either code for amino acids, and makes up the 3'untranslated region (Fig. 31-2, green 3'). A codon or triplet code is determined by a base triple (three contiguous nucleotides). A codon provides information about a certain amino acid to be added into the peptide chain or for the chain to stop. Three codons in mRNA (UAA, UAG and UGA) are stop codons (Fig. 31-2). The signals encoded by the codon form the genetic code. The amino acids are carried on small RNA molecules called transfer RNA (tRNA). Each of these tRNA molecules contains an anticodon consisting of 3 unpaired nucleotides, which carry complementary bases to the bases on the mRNA. Additionally, each tRNA carries a specific amino acid for the prolonged polypeptide. There are at least 20 different types of tRNA, each carrying only one amino acid. Ribosomal RNA (rRNA) molecules diffuse from the nucleus to the cytoplasm and into the ribosomes, where it plays an essential role in the protein synthesis (Fig. 31-3). Every anticodon or base triplet recognizes its complementary codon in the mRNA. The tRNA deposits its amino acid in the peptide chain.
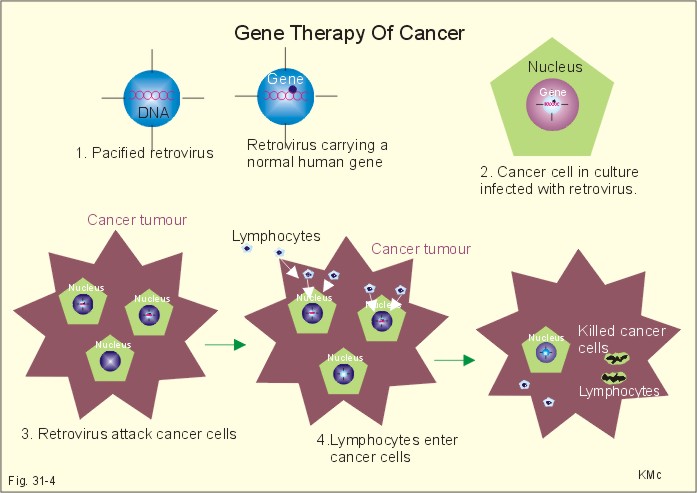
Fig. 31-3: Increased protein copying in the ribosomes - ribosomal translation. In the cells the nucleotide strands of DNA are separated bit by bit and a special enzyme, DNA polymerase, synthesizes new DNA. DNA replication is normally an accurate process at which exact copies of the DNA molecule are made. Still, a 10-30 errors occur in the DNA replication of each body cell every day. Fortunately, efficient gene reparation processes correct the majority of these errors. This paragraph deals with 1. Mutations, 2. Genetic disorders, 3. Gene therapy, and 4. Other strategies. Rarely occurring errors in DNA replication are called mutations. Several types of mutation occurs: 1.1. Termination mutations. Mutations involving stop codons affect the normal polypeptide chain termination. A nucleotide change allows the insertion of an extra amino acid before termination, or delete one amino acid from the normal sequence. A typical example is the rare type of a-thalassaemia called Haemoglobin constant Spring. The globin part of normal haemoglobin consists of two a- and two b-chains. In contrast, mutations in the stop codon produce a-chains with many extra amino acids in thalassaemia. Thalassa means sea and originally referred to the distribution of this particular anaemia along the coastline of the Mediterranean Sea. Today thalassaemia is found everywhere. 1.2. Splicing mutations. When mutations occur in the DNA sequence, which normally code the splicing enzymes and thus direct the splicing of introns from RNA, the result may be abnormal splicing with introns in the final mRNA. Such a mRNA is translated into a protein molecule, which is abnormal because there is an erratic amino acid in the polypeptide chain. 1.3. Insertion-deletion mutations. Insertion or deletion of one or more nucleotides in DNA by mutation results in an abnormal sequence. The gene for a-chains in haemoglobin is duplicated on both chromosomes 16. Mutations may result in deletion of one or both genes on each chromosome 16. When all four a-chain genes are absent, the a-chain synthesis is impossible and only gamma 4-chains are present. These chains cannot carry oxygen, and infants are stillborn or die shortly after birth (the new-born is pale and oedematous: hydrops foetalis). When three genes are deleted, there is anaemia called a-thalassaemia. When only one or two genes are deleted there is a microcytosis and polycythaemia called a-thalassaemia traits (ie, essentially healthy carriers). Possession of an angiotensin converting enzyme gene deleter (ACE-D), which delete a 287 bp repeat sequence in the enzyme, results in high concentrations of circulating enzyme, and a significantly higher frequency of myocardial infarction in genotype DD. Deletions in the dystrophin gene of the X chromosome remove coding sequences, so the muscles are deprived of the cytoskeletal muscle protein, dystrophin. Dystrophin is the normal gene product, which is normally linked to the sarcolemma as a network to the sarcomers. Lack of dystrophin is the cause of Duchenne Muscular Dystrophy (DMD). DMD is a X-linked recessive disease caused by the defect dystrophin gene. DMD also occurs spontaneously by mutation in the DMD locus on the X chromosome (ie, the Xp21 region). There is proximal limb weakness with pseudohypertrophy of the calves. The suffering boys have to climb up their legs with their hands, and they have difficulties in walking and running. The creatine phosphokinase concentration in the plasma is substantially elevated. Muscle biopsies show phagocytosis, fibre necrosis, and absence of dystrophin, regeneration, and fatty patches. Exercise helps to preserve muscle function by activation of otherwise inactive synergists. The boys usually die before adolescence (20 years of age). 1.4. Point mutations. Substitution of one nucleotide for another may totally change the codon in a coding sequence. Carcinogens can cause point mutations in genomic DNA, and if the mutations occur in the coding region they may be pathogenic. In sickle cell anaemia a mutation in the b-chain gene, changes the codon from GAG to GTG. The codon GAG initiates translation of a polypeptide chain with glutamic acid and the codon GTG incorporate valin in each of the two b-chains. The product is a highly unstable b-chain, which cannot be utilised for oxygen transport. This particular haemoglobin is called Haemoglobin S or Sickle cell Haemoglobin. Exposed to low oxygen tensions these molecules form elongated crystals inside the red cells. The spiked ends of the crystals rupture the cell membrane leading to haemolysis and sickle cell anaemia. Sickle cell anaemia is the homozygous state, where both genes are abnormal (HbSS), whereas sickle cell trait is the heterozygous state (HbAS), with only one chromosome carrying the abnormal gene. The disease (HbSS) manifests itself at about 6 months of age, where the concentration of haemoglobin F decreases towards adult levels. Different degrees of b-thalassaemia also occur following point mutations. The b-chains are not produced or produced to a limited extent, whereas there is an excess of a-chains. The precipitation of a-chains causes haemolysis of erythroblasts and erythrocytes and inefficient erythropoiesis. Homozygous b-thalassaemia cases have no b-chains (bo) or too few (b+). In heterozygous b-thalassaemia there may be a mild anaemia and usually symptomless microcytosis. Genetic disorders are classified into 2.1. chromosomal, 2.2. Single gene defects, and 2.3. Multifactorial disorders When a chromosome fail to separate during meiosis, the ovum or sperm gets an extra chromosome and become trisomic, or no chromosome and become monosomic. Sex chromosome trisomy (XXY) is called Klinefelter's syndrome. Sex chromosome monosomy (only one X and no Y) is called Turner's syndrome (Chapter 29). 2.2. Single gene defects. Each diploid cell contains two copies of all autosomes. If one of the two copies has a mutation and the normally produced protein cannot compensate, then an autosomal dominant disorder occurs. Examples are achondroplasia, porphyria, a1-antitrypsin deficiency, Huntingtons's chorea, and von Willebrand's disease. When both chromosomes carry the mutated gene, an autosomal recessive disorder appear. This is not the case when only one mutated gene is present. These patients are heterozygous for the mutated gene and thus unaffected healthy carriers. Examples are mucoviscidosis or cystic fibrosis, Wilson's disease, and many other inborn errors of metabolism. Pancreatic cystic fibrosis (mucoviscidosis) This is a recessive genetic defect with dysfunction of exocrine glands.Cystic fibrosis is an autosomally recessive genetic disorder caused by a cystic fibrosis gene. There is a gene mutation in chromosome 7, which result in a defect in a regulator protein (Cystic Fibrosis Transmembrane Conductance Regulator, CFTR) - a defect b-adrenergic gated chloride-channel. The defective chloride-channel fails to open in response to an increase in intracellular cAMP in the pancreatic ducts, the airways and the sweat glands. The patients have a minimal chloride excretion. The decreased excretion of chloride and increased reabsorption of Na+ and water produce a small viscoid secretion that closes and dilatates the duct systems. Finally, the ducts are destroyed (ie, chronic respiratory disease and pancreatic insufficiency). Fully manifested cases suffer from defect mucous secretion (mucoviscidosis) with chronic respiratory disease, cystic pancreatic fibrosis with pancreatic insufficiency, and abnormally high [NaCl] in sweat. This life-threatening condition is thus a genetic defect in the b-adrenergic-gated Cl--channels of the glands in the airways, the pancreas, and in the sweat glands. Bronchopulmonary disorders result in chronic hypoxia with finger clubbing. Pancreatic failure with lack of digestive enzymes results in steatorrhoea and cholesterol gallstones. A sweat test resulting in a Na+-concentration above 60 mM in the sweat is strongly indicative of cystic fibrosis. Cystic fibrosis is treated with amiloride, which block the ductal Na+- reabsorption, or with ATP, which stimulates Cl--secretion by a pathway different from the missing cAMP. In some cases of cystic fibrosis, with severe pulmonary insufficiency, lung transplantations have been performed successfully. Albinism (AMELANOSIS) is inherited as an autosomal recessive disorder of melanin synthesis. The biosynthesis of the enzyme tyrosinase is defective, which results in lack of melanin. Amelanosis is manifest by white hair, pink-white skin, blue eyes and photophobia. Phenylketonuria (PKU) is also an autosomal recessive disorder. There is a defect conversion of phenylalanine to tyrosine and thus hypopigmentation. The genetic defect results in lack of the enzyme phenylalanine 4-hydroxylase. PKU must be diagnosed and treated soon after birth in order to avoid severe mental retardation. PKU patients almost never reproduce. PKU occurs once in 25000 live births in the population. Carbohydrate malabsorption: The most common chronic disorder in humans is lactose malabsorption or hypolactasia (lactose-induced diarrhoea or lactose intolerance), which is due to a genetically deficiency of lactase in the brush-border of the duodeno-jejunal enterocytes. More than 50% of all adults in the world are lactose intolerant. Infants with the rare congenital lactase intolerance are borne without lactase in their brush border. They develop diarrhoea, when they are breast-fed. This can result in a life threatening dehydration. The amount of lactose entering the colon determines the size of the osmotic diarrhoea. Milk made from Soya beans and fructose is well tolerated. Sucrase-isomaltase deficiency is an autosomal recessive genetic condition with sucrose intolerance. This disorder is found in 10% of Eskimos (Inuits), which is not surprising, since they must have lived for thousands of years of a natural diet without sucrose. Glucose-galactose malabsorption is a rare genetic disorder caused by a defect in the brush border system for glucose and galactose absorption (GLUT-5). Fructose is well tolerated. 2.3. Multifactorial gene defects. These disorders involve many genes and often also environmental factors. Examples are congenital pyloric stenosis, asthma, hypertension, schizophrenia, congenital heart disease and genetic cancer. Cancer is often multifactorial. Cancer arises from only a single cell, which suddenly starts to proliferate out of control. Oncogenes encode proteins that normally are involved in cellular proliferation and enhance cellular growth. Tumour suppressor genes suppress undue cellular proliferation. Mutations in the normal RB gene result in retino-blastoma. Mutations in the normal p53 gene may lead to brain tumours, carcinomas of the breast and lungs, osteosarcomas, colon carcinomas and leukaemia. A nuclear phosphoprotein (53-kDa) encoded by p53, is involved in DNA repair and synthesis. In many cases of different cancer types there is a secondary mutation of the p53 allele. Gene therapy is the application of gene transfer (RNA or DNA delivery to cells) in an attempt to treat genetic or acquired disorders. The transferred DNA must contain promoter zones for transcription and the protein-coding region of the gene. Insertion of DNA into cells is performed with a large number of techniques, among which plasmid-based (plasmid mixed with lipid micelles), and virus-based (retrovirus, adenovirus, herpes simplex virus) vectors are the most effective. The most widely used vehicle is genetically engineered retrovirus from the Moloney murine leukaemia retrovirus. Retroviruses can cause cancer, when they infect a cell and activate resting or proto-onchogenes. Retrovirus may also be used in future gene therapy, as described in the following: 1. A normal gene, which can activate the immunodefence system, is placed inside a manipulated, pacified retrovirus (Fig. 31-4).
Fig. 31-4: Gene therapy of cancer. 2. Cancer cells, removed from the tumour of a patient, are cultured in vitro and infected with the retrovirus carrying the normal gene. 3. Selected cells from the culture are implanted in the cancer tumour of the patient, where the retrovirus attacks some of the cancer cells. 4. The immune-activating genes signal to the immune defence system to forward lymphocytes. The lymphocytes enter the cancer cells and destroy them (Fig. 31-4). 4. Other strategies are the following: Retrovirus is also used for gene transfer of the cytokine, interleukin-2 and of tumour necrosis factor, in attempts to support the immune response to cancer. Bone marrow cells from a patient with b-thalassaemia are infected with a pacified retrovirus carrying a normal human b-globin gene. Hereby, cells capable of normal erythropoiesis can replace a sufficient number of abnormal bone marrow cells. Low density lipoprotein (LDL) receptor deficiency causes hereditary hypercholesterolaemia. Insertion of the normal gene for the LDL- receptor into the patients’ abnormal hepatocytes may have beneficial effect. Gene transfer can also result in the production of a cell membrane protein, such as the chloride-channel transmembrane regulator gene that has mutated in cystic fibrosis. This is transferred to pulmonary cells by aerosol technique or by vectorial insertion into the target cells. Equation for Gene Frequency Calculation In the absence of mutation and random genetic drift, genotypes in a population of random mating will be given by the equation: Eq. 31-1: (p + q)2 = 1 or expanded: (p2 + q2 + 2pq) =1, where p is the frequency of the normal gene in the population, and q is the frequency of the abnormal gene. Obviously, p2 is the frequency of the normal homozygote, q2 is the frequency of the abnormal homozygote (all affected by the abnormality), and 2pq is the frequency of healthy carriers. Note that (p + q) is 1. This is the Hardy-Weinberg theorem, which is used to calculate the frequency of abnormal genes in the total gene pool of a population. Each of the following 5 statements have True/False options: A. Phenylketonuria (PKU) is an autosomal dominant disorder. B. Inside the ribosome the particular sequence of the mRNA is read, and the particular sequence of nucleotides is build into a polypeptide. C. Sex chromosome trisomy (XXX) is called Klinefelter's syndrome. D. When carcinogens cause point mutations in genomic DNA inside the coding region, they are often pathogenic. E. The first nucleotides on the mRNA form a regulatory sequence is called the 5’untranslated region, and does not code for amino acids. A female and a male, who plan to have children, want advice concerning a certain genetic disease. The incidence of the recessive disease is 10-4 in the population. What is the frequency of the recessive gene occurring in one human being? A newborn girl suffers from almost continuous coughing. The mother contacts her doctor, who - at the third consultation - suspects cystic fibrosis and arrange a sweat test to be performed. Pilocarpine iontophoresis facilitates the collection of sweat. The NaCl concentration in sweat is found to be 70 mM, which is several fold the normal value. The patient suffers from bronchopulmonary infection with mucoviscidosis of the exocrine gland ducts, malabsorption, fatty stools (steatorrhoea), and deficiency of fat soluble vitamins (A, D, K). - The incidence of cystic fibrosis is approximately 1 out of 1600 live births. 1. What is cystic fibrosis? 2. Calculate the frequency of the abnormal cystic fibrosis gene. 3. Calculate the frequency of the normal homozygote. 4. Calculate the frequency of the heterozygous carrier of cystic fibrosis. A female, whose father is an albino, plan to marry a male albino (genotype aa). They wish to know the probability of having albino children and albino carriers. 1. What is albinism? 2. What are the probability of having an albino child? 3. What are the probability of having albino carriers among their children? The brother of a Phenylketonuria (PKU) patient seeks genetic advice before marriage. The brother is normal and cases of PKU are excluded for generations in the family of the female. 1. What is PKU? 2. What are the probability that the brother is heterozygous or normal? 3. What are the probability of the couple having a PKU child? 4. PKU patients rarely reproduce. How can the gene persist in the population? A 3-year-old boy has difficulty in standing and walking. It is particularly difficult for him to reach the standing position from the floor, and he has to climb up his legs with his hands. The thighs of the boy are abnormally thin, but the calves are muscular and almost hypertrophic. The creatine phosphokinase concentration in the plasma is 100 times the normal level. – The mother of the boy had a brother, who died at the age of 20 by the same disorder. 1. What is the diagnosis? 2. Why are the creatine phosphokinase in plasma elevated? 3. Why are such disorders confined to boys? 4. Explain the hypertrophy of the calves. 5. What are the risk that the sister to a boy with this muscular disease gets a child with manifest muscular disease? Try to solve the problems before looking up the answers. · Nuclear DNA is the dominant controller of protein synthesis in the ribosomes. The codes of special enzymes and other proteins are based on at least 20 amino acids arranged in sequence. · The nucleus contains a nucleolus and the chromatin network. Chromatin is DNA molecules rolled up in a pearl chain structure with proteins called nucleosomes. · DNA consists of two nucleotide strands twisted around each other to form the so-called double helix, which can be several cm in length. The double helix is folded to form chromosomes with a length of approximately 10 mm. · The human body cell contains 46 chromosomes in a normal nucleus. Females have two X-chromosomes and two sets of 22 autosomes, whereas males have one X- and one Y-chromosome plus two sets of 22 autosomes. · The entire human genome on the 24 different chromosomes is estimated to be 3 109 bp long and to contain 105 genes. · Dystrophin is the normal gene product, which is normally linked to the sarcolemma as a network to the sarcomers. Lack of dystrophin is the cause of Duchenne Muscular Dystrophy (DMD). DMD is a X-linked recessive disease in boys caused by the defect dystrophin gene. · Sickle cell anaemia is the homozygous state, where both genes are abnormal (HbSS), whereas sickle cell trait is the heterozygous state (HbAS), with only one chromosome carrying the abnormal gene. The disease (HbSS) manifests itself at about 6 months of age, where the concentration of haemoglobin F decreases towards adult levels. · Homozygous b-thalassaemia have no b-chains (bo) or too few (b+). In heterozygous b-thalassaemia there may be a mild anaemia and usually symptomless microcytosis. · Each diploid cell contains two copies of all autosomes. If one of the two copies has a mutation and the normally produced protein cannot compensate, then an autosomal dominant disorder occurs. Examples are achondroplasia, porphyria, a1-antitrypsin deficiency, Huntingtons's chorea, and von Willebrand's disease. · When both chromosomes carry the mutated gene, the autosomal recessive disorders appear. This is not the case when only one mutated gene is present. These patients are heterozygous for the mutated gene and thus unaffected healthy carriers. Examples are mucoviscidosis or cystic fibrosis, Wilson's disease, and many other inborn errors of metabolism. · Pancreatic cystic fibrosis is a recessive genetic disease caused by dysfunction of exocrine glands. The defect is in a transmembrane regulator protein called the cystic fibrosis transmembrane conductance regulator (CFTR). The CFTR represents a b-adrenergic gated chloride channel, which is normally opened by elevated intracellular cAMP. · The cystic fibrosis patients have a minimal chloride excretion and thus as minimal excretion of salt and water into the duct systems. This is what makes all exocrine secretions viscid, the duct systems are occluded and dilatated; finally the ducts are destroyed (ie, chronic respiratory disease and pancreatic insufficiency). · Albinism (AMELANOSIS) is inherited as an autosomal recessive disorder of melanin synthesis. The biosynthesis of the enzyme tyrosinase is defective, which results in lack of melanin. Amelanosis is manifest by white hair, pink-white skin, blue eyes and photophobia. · Phenylketonuria (PKU) is also an autosomal recessive disorder. There is a defect conversion of phenylalanine to tyrosine and thus hypopigmentation. The genetic defect results in lack of the enzyme phenylalanine 4-hydroxylase. PKU must be diagnosed and treated soon after birth in order to avoid severe mental retardation. PKU patients almost never reproduce. PKU occurs once in 25 000 live births in the population. · Multifactorial gene defects. These disorders involve many genes and often also environmental factors. Examples are congenital pyloric stenosis, asthma, hypertension, schizophrenia, congenital heart disease and cancer. · Genetic cancer is often multifactorial. Cancer arises from only a single cell, which suddenly starts to proliferate out of control. Oncogenes encode proteins that normally are involved in cellular proliferation and enhance cellular growth. · Tumour suppressor genes suppress undue cellular proliferation. Mutations in the normal RB gene result in retino-blastoma. · Gene transfer can result in the production of a cell membrane protein, such as the chloride-channel transmembrane regulator gene that has mutated in cystic fibrosis. This is transferred to pulmonary cells by aerosol technique or by vectorial insertion into the target cells. Nature. Weekly journal published by Macmillan Magazines Ltd, Porters South, 4 Crinan Street, London N1 9XW, UK. Jorde LB, Carey JC, Bamshead MJ and RL White. Medical Genetics. 4th Ed. Mosby, St Louis, 2009, Scientific American. - Example: Verma, IM: Gene therapy. Sci Am pp 68-72, 81, 82, 84. Nov 1990.
|
||
.jpg){kind=link}
Click here to introduce your comments or contributions