New Human Physiology | Paulev-Zubieta 2nd Edition
Chapter 16: Control of Respiration - High Altitude
| HOME | PREFACE | TABLE OF CONTENTS | SYMBOLS | SECTION INFO | CONTRIBUTORS | LINKS | CONTACT US |
Highlights
Study_ObjectivesPrinciplesDefinitionsEssentials
PathophysiologyEquationsSelf-AssessmentAnswers
Further Reading
|
Chapter 16
|
 |
|
|
· To define the following concepts acidaemia, blood-brain-barrier, and different types of hypoxia. · To describe respiratory centres, apneustic and pneumotaxic centres, central & peripheral chemoreceptors, bronchopulmonary stretch receptors, irritant-receptors, J-receptors, slow-receptors, and other receptors affecting cardiopulmonary control. · To draw ventilatory response curves to carbon dioxide at sea level, during sleep, at altitude, during exercise and acidosis. · To calculate gas tensions and ventilatory variables from relevant variables given. · To explain the respiratory autonomy, CNS-effects, adaptation to altitude- hypoxia – hypercapnia –exercise - acid-base disorders. To explain mountain sickness, foetal oxygenation and cardiopulmonary changes at birth. · To use the above concepts in problem solving and case histories. · Dalton´s law: The partial pressure or tension of a single gas in a mixture is equal to the product of the total pressure and the mole fraction (F). · Acidaemia is acid poisoning with a pH below 7.35 in the arterial blood. · Apneusis is a particular respiratory pattern with prolonged inspirations separated by irregular short expirations, following midpontine transection when the vagi are cut. · Blood-brain-barrier consists of tight junctions between the endothelial cells of the capillaries in the CNS. The tight junctions allow only small molecules to pass into the brain tissue, and they are impermeable to H+ and HCO3-, whereas lipid-soluble gasses such as carbon dioxide can pass the plasma membrane. · Blood-CSF-barrier is a tight junction barrier across the chorioid plexus, preventing many large molecules to pass from the blood to the cerebrospinal fluid (CSF). · Carotid bodies are small organs located between the internal and external carotid arteries. The bodies contain chemosensitive glomus cells, fixed in a plasma-like tissue-fluid surrounded by abundant sinusoidal capillaries. · Circumventricular organs are discrete structures in the hypothalamus, the pineal gland and the area postrema, with highly fenestrated capillaries that can be easily penetrated by large and small molecules as well as ions. The circumventricular organs are located close to essential control centres regulating respiration, blood-glucose level, and the osmolality of the extracellular fluid. · Hypoxaemia refers to a condition with low oxygen concentration in the arterial blood including hypobaric or hypotonic hypoxia. · Medullary, central chemoreceptors are located below the ventrolateral surface of the medulla and communicate with the respiratory centre neurons. The main stimulus of these receptors is the hydrogen ion concentration of the brain extracellular fluid, and they respond to acidaemia with hyperventilation. · Polycythaemia refers to a condition with an increase in red cell count, blood haemoglobin and packed cell volume (PCV or haematocrit). Polyerythrocythemia refers to a sign of disease, with an increase in red cell count, blood haemoglobin and haematocrit (above the normal high altitude levels). It does not include Polycythemia Vera, where there is also an increase of white blood cells and platelet count, never seen at high altitude by the authors.
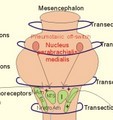
This paragraph covers 1. Neural and 2. Chemical control of breathing, 3. Acclimatization, 4. Alveolar gas oscillations, and 5. Oxygen-altitude-Mt. Everest. 1. The neural control of respiration involves three components: I. The central neurons, II. Effectors, and III. Sensors. Results of brainstem transections performed on animals over the past two centuries indicate that the respiratory controller or respiratory centre (RC) resides in the brainstem. Such transection results also imply the presence of a pneumotactic off-switch centre in the nucleus parabrachialis of the rostral pons (Fig. 16-1). RC is a collective term for a diffuse network of at least two types of neurons located in the medullary reticular formation. The inspiratory I-neurons fires just before and during inspiration, and they are tonically active in providing inspiration. The expiratory E-neurons fire just before and during expiration. I- and E-neurons are localized in at least two medullary groups, the dorsal and the ventral motor groups. The dorsal motor group in the solitary tract nucleus contains mainly I-neurons. The axonal projections of these neurons terminate in the cervical and thoracic spinal motor neurons of the phrenic and intercostal nerves. Nucleus tractus solitarius is also concerned with cardiovascular control. The ventral motor group resides in the nucleus ambiguus, para-ambigualis and retro-ambigualis (ie, the Bøtzinger complex). These diffusely located neurons include both I- and E-neurons, and their E-neurons project to expiratory intercostal motor neurons in the thoracic segments. The I-neurons show bursts of spontaneous activity (approximately 12-16 times per min) separated by quiet periods, in a pattern mimicking the breathing at rest. The E-neurons, on the other hand, are not self-excitatory. The activity of the I-neurons correlates with the rate and depth of breathing. The basic activity of the I-neurons, like that of all pacemakers, is modulated by a multitude of external signals from the periphery and from other areas of the CNS. The central inspiratory drive determines the intensity of the desire to inspire. This drive is measured as the inspiratory flow rate or as the phrenic nerve activity. The phrenic nerve innervates the diaphragm, which is the most important inspiratory muscle. The duration of the central inspiratory drive determines the inspiratory time (ie, the active phase of medullary pacemaker I-neurons). Inspiratory neural activity is terminated by either impulses from the pontine off-switch or from the pulmonary stretch receptors conducted through the vagal nerve. Inspiratory airflow equals driving pressure divided by resistance. Accordingly, the inspiratory flow rate is a function of the pressure- generating respiratory muscles. The smooth muscles of the upper airways determine upper airway resistance. The expiratory phase lasts as long as the inspiratory off-switch neurons are active, and inspiration is not resumed. Expiration is passive, except at very high levels of ventilation, where expiratory muscles contract rhythmically to augment expiration. The main stimulus to breathing is CO2 through the medullary chemoreceptors, located close to the RC. Normal respiratory rhythm (eupnoea) can persist after removal of the entire brain above the brainstem (ie, in a decerebrated animal). This confirms the presence of a basic inspiratory tonic activity. Transection made at the midpons, with the vagi intact, cause slowing and deepening of respiration. When the vagi are also sectioned, midpontine transection results in apnoea (Fig. 16-1) or apneusis (ie, prolonged inspirations or I-spasms, separated by short expirations). Removal of the pontine centres by transection between the pons and medulla results in a gasping, irregular pattern called ataxic ventilation (Fig. 16-1). The neurons of the medulla themselves have a spontaneous rhythmicity. Thus, the role of the pontine centres is to make the discharges of the medullary neurons smooth and regular. Transection between the medulla and the spinal cord results in respiratory arrest (apnoea). Respiratory rhytmogenesis is most likely due to a central pattern generator located in the brainstem and operating as described above (Fig. 16-1). A network of interneurons in the brainstem is termed the reticular activating system, because it affects our state of wakefulness and increase respiratory activity.
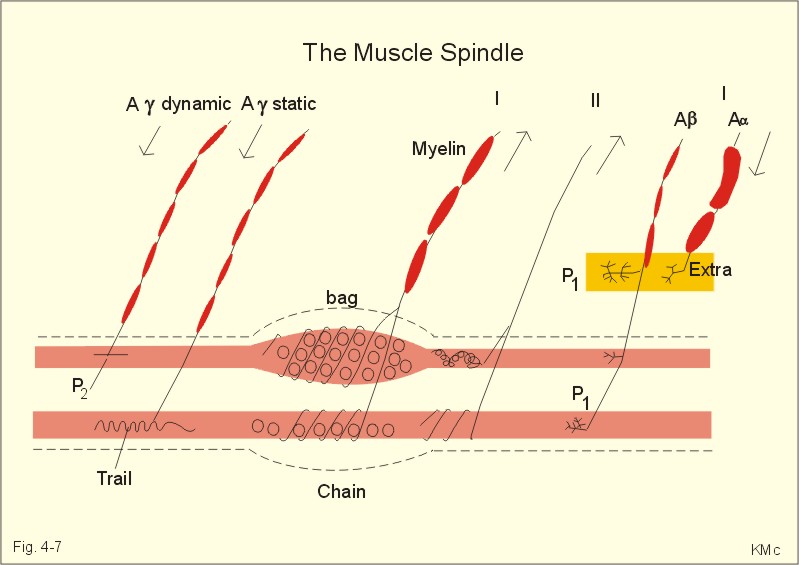
Fig. 16-1: The respiratory rhythm generator with results of brain stem transection. NTS = nucleus tractus solitarius, NAm = nucleus ambigualis, NretroAm= nucleus retroambigualis. The descending brain impulses that drive the cervical and thoracic motor neurons consist of those arising from voluntary and involuntary cerebral sources. These motor neurons in cranial and spinal nerves lead to the respiratory muscles and to the smooth muscles of the upper airways. The corticospinal tract transfers voluntary commands directly from the cortex to the somatic motor neurons, or passes the same information through the pontomedullary centres before descending to the motor neurons via the reticulospinal tract. The involuntary descending signals arise from the I-neurons in 3 brainstem nuclei. These are the solitary tract nucleus (via the phrenic nerve to the diaphragm), the mixed neurons in the nucleus para- and retro-ambigualis (via the intercostal nerves to the intercostal muscles), and the nucleus ambiguus (via the cranial nerves to the airways including smooth muscles). The intrafusal muscle fibres can shorten relatively more than the extrafusal (Fig. 4-7). Hence, a simultaneous gamma- and a- efferent discharge shortens the respiratory muscles to provide exactly the necessary power. The respiratory muscles (except the diaphragm) show this so-called gamma-loop servo. Actually, this is a sensory feedback to ensure homeostasis, but we must look at other sensors for the control system. 1. Higher brain centres (cortex, hypothalamus, and diencephalon). Cortical, voluntary breath holding is possible until the breaking point is reached (ie, the point where apnoea is disrupted). Similar responses are released by stimulation of the diencephalon. A rise in hypothalamic temperature triggers frequent breathing (tachypnoea) via the respiratory centres. During sleep and anaesthesia, breathing provides for metabolic needs primarily via the automatic homeostatic control system described above. During wakefulness, however, the breathing system serves both homeostatic and voluntary, behavioural (nonhomeostatic) needs. The behavioural needs include sucking, swallowing, speech, singing, laughing, crying, defaecation, voiding, breath holding, exercise, hyperventilation, and coughing. Such voluntary acts affect PaCO2, PaO2, and [H+]. 2. Bronchopulmonary stretch receptors are located in the smooth muscles of the trachea, larger bronchi and also in the lung parenchyma in the alveolar ductules and sacs. The activity of these smooth muscle receptors increases markedly with airway distension, and the activity ebbs slowly with time, hence they are called slowly-adapting pulmonary receptors. If the tidal volume exceeds one litre, these receptors initiate signals that inhibit the inspiratory drive via myelinated vagal fibres, reinforcing the actions of the pontine centres and protecting the lungs from overexpansion. In humans this Hering-Breuer reflex plays no part in regulating ventilation during quiet breathing at rest, but the reflex is active during exercise. 3. Rapidly adapting irritant receptors are probably free or modified vagal nerve endings in the epithelium of the airways. These receptors are stimulated by irritants (smoke, allergens) and by inflammatory mediators such as prostaglandins and histamine. Rapidly adapting irritant receptors mediate the protective and sometimes pathologic responses of cough and bronchospasm. The efferent limb of this reflex is the motor fibre in the vagus, which initiates bronchospasm. 4. Juxta-pulmonary capillary receptors are terminals of non-myelinated vagal fibres (J-receptors). Distension of the interstitial space, as seen in a variety of cardiopulmonary disorders increases the diffusion distance, elicits increased ventilation, tachypnoea, bradycardia, and low arterial pressure via vagal reflexes. These cardiopulmonary disorders are microemboli, pulmonary oedema, pneumonia, fibrosis, atelectasis, and damage from inhaled irritants. Atelectasis means alveolar collapse. 5. Peripheral arterial chemoreceptors are found in the carotid bodies of humans (the aortic bodies account for only a small effect). The carotid chemoreceptors transmit impulses to CNS via cranial nerves IX and X, and induce rapid changes in ventilation. The glomus cell is sensitive not only for low PaO2 (hypoxaemia), but also for increasing PaCO2 (hypercapnia), hyperkalaemia and acidosis. 6. Central, medullary chemoreceptors are essential and particularly important to steady state ventilation. 7. Thermoreceptors, stimulated by a rise in core temperature (so-called heat receptors), elicit tachypnoea and tachycardia (frequent heart rate) via hypothalamus. Other thermoreceptors termed cold receptors are stimulated by a fall in shell temperature. Stimulation of these receptors elicits bradycardia via the regulating hypothalamic temperature area. This so-called survival reflex (or “diving bradycardia”) protects the bloodflow through the brain, heart, and lung in emergency situations involving breath holding, not only in water, but also on land. 8. Arterial baroreceptors in the carotid sinus are stretch receptors, stimulated by increased transmural arterial pressure. Stimulation decreases arterial pressure, and inhibits heart rate and ventilation as in the survival reflex. 9. Receptors in working muscles stimulate ventilation via type III and IV afferents to the respiratory centres in the medulla. These receptors are involved in the exercise hyperpnoea. 10. Protective, vagal reflexes are related to vomiting, hick-up and swallowing. These reflexes protect us from inhaling vomit and thus being choked. 2. The chemical control of respiration The respiratory system exerts its homeostasis by both peripheral arterial and by central medullary chemoreceptors. Both types of receptors are sensitive to changes in the proton concentration ([H+]) around them. Such changes imply changes in the intracellular [H+], and changes in the ionic composition (Na+, K+, and Ca2+). Both types of receptors are activated by increases in PCO2 independent of [H+]. Acute hypoxia stimulates the peripheral arterial chemoreceptors, but maintained hypoxia has a depressant effect on both central chemoreceptors and regulatory neurons in the RC. The carotid and aortic bodies (glomera carotici & aortici) are small organs located in the tissue between the internal and external carotid arteries and at the arch of the aorta, respectively. The bodies contain chemosensitive glomus cells, fixed in a plasma-like tissue fluid surrounded by lots of sinusoidal capillaries, which have an extremely large flow (up to 2000 ml *min-1*100g-1) of arterial blood.
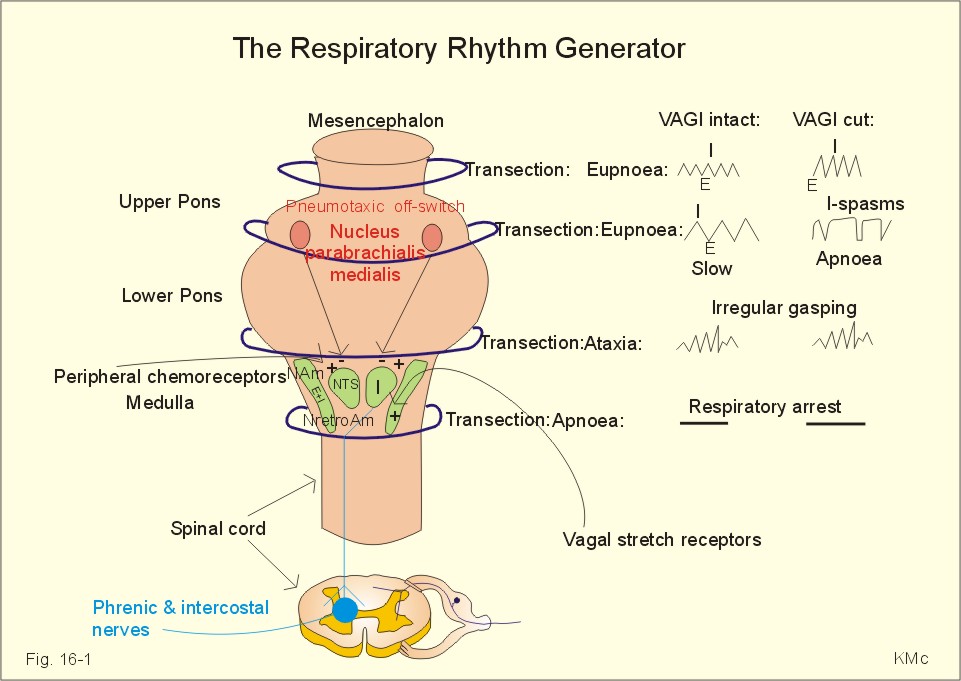
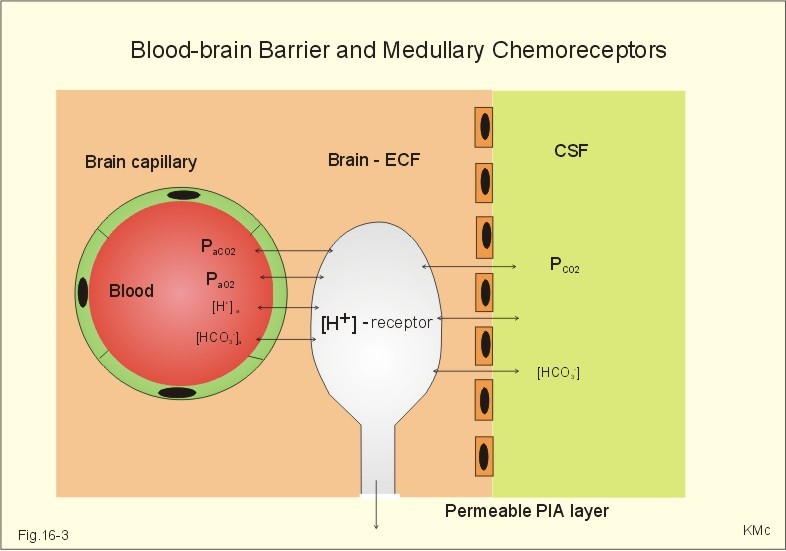
Fig. 16-2: Glomus cell with a rich arterial blood supply. The IXth cranial nerve is from the carotid bodies and the Xth (vagus) nerve from the aortic bodies. The frequency of action potentials in the glomus nerve increases, when PaO2 is below normal. Glomus cells (type I) are sensitive to decreased PaO2 (hypoxaemia mediating hyperventilation), but also to increased PaCO2 (hypercapnia) and increased [H+] (acidaemia), and [K+] (hyperkalaemia) in the arterial blood and thus in the fluid surrounding the glomus cells (Fig. 16-2). The carotid bodies are predominant in humans. The carotid sinus nerve innervates these bodies, which is a branch of the glossopharyngeal nerve (IX). The following hypothesis explains how the 4 glomus cell stimuli operate. Normally, an oxygen-sensitive Na+-K+-pump (Skou´s enzyme) maintains the high intracellular K+ and the low intracellular Na+ of the glomus cell. Hypoxia inhibits this pump, whereby the cell membrane depolarises. The stimulation by hyperkalaemia is an unspecific depolarisation, which occurs in most cell membranes including the glomus cells. Hypercapnia and acidaemia feed the cell with CO2, which rapidly converts into H+ and HCO3-. Increased acidity in the cell stimulates the H+-Na+–exchanger in the cell membrane, the Na+-K+-pump is inhibited and depolarisation occurs. Depolarisation triggers a voltage-gated Ca2+-channel. The high Ca2+-influx activates neurosecretory granules and releases an excitatory neurotransmitter that depolarises the nerve fibre membrane and a propagating signal for the RC is transmitted (Fig. 16-2). The glomus cell contains lots of the neurotransmitter, dopamine, and the enzyme, carboanhydrase. - However, the determinant of the signaling cascade is actually unknown and the mechanism for hypoxia and hypercapnia are probably different. The short latency and response times of the carotid chemoreceptors and the location of the receptors close to the heart and lung, means rapid ventilatory stimulation. The carotid bodies are therefore responsible for the immediate reactions to changes in PaO2, PaCO2, [H+], and plasma-[K+]. Stimulation of the carotid bodies by hypoxaemia, hypercapnia, hyperkalaemia, and acidaemia increases the impulse frequency of the carotid sinus nerve and the glossopharyngeal nerve to the medullary respiratory centres (RC). Increased activity from RC to the respiratory striated muscles and the upper airway smooth muscles causes increased ventilation. This has a homeostatic effect on the initial stimuli - partially relieving hypoxaemia, hypercapnia and acidaemia. This is a negative feedback loop. Falling PaO2 stimulates the carotid glomus cells to increase ventilation (Fig. 16-2 right), but the hyperventilation simultaneously reduces PaCO2, which counteracts the stimulation of breathing. Patients with a PaO2 below 7.3 kPa are hypoxic and suffer from hypotonic or hypobaric hypoxia. Compensatory hyperventilation maintains adequate oxygenation in the face of acute decreases of PIO2 from the normal 20 kPa (150 mmHg), until PIO2 decreases below 4 kPa (30 mmHg), where consciousness is lost. There is an inverse or hyperbolic relationship between alveolar ventilation and PaO2. At the peak of Mt. Everest the PaO2 is just below 4 kPa in trained mountain climbers, and they can walk only with great difficulty. Anaemic patients have reduced oxygen content in the arterial blood (CaO2) in spite of normal PaO2. Since PaO2 represents the stimulus to the glomus cells of the carotid bodies, and not the oxygen concentration of the arterial blood, such conditions are rarely associated with compensatory hyperventilation. A person with CO poisoning can die without increasing his breathing and without having felt shortness of breath (dyspnoea). CO acts as metabolic poison at the carotid bodies and increases ventilation to some extent. Persons without peripheral arterial chemoreceptors can still increase their ventilation, in response to increases in PaCO2 or [H+]. These two stimuli have a particularly strong effect on the central, medullary chemoreceptors. Three superficial (subpial) areas have been defined on the ventrolateral surface of the medulla, designated L (Loeschke), M (Mitchell) and S (Schläfke) after the scientists that located the medullary chemoreceptors. Areas L and M are believed to be chemosensitive, and nerve fibres from L and M converge on area S, where they pass deeper into the medulla to reach the medullary Respiratory Centre (RC). The medullary chemoreceptors are located 100-200 mm below the ventrolateral surface. These receptors communicate with RC neurons located deeper in the medulla. The main stimulus to the chemoreceptors is the [H+] of the brain extracellular fluid (ECF), which is in close proximity to the cerebrospinal fluid (CSF) that bathes these receptors. The thin layer of pia between CSF and the brain ECF is highly permeable to CO2, and even to ions, whereas the blood-brain barrier resists ionic diffusion (Fig. 16-3). It takes several minutes for changes in blood [H+] to be reflected in the brain ECF and hours for equilibrium. The tight junctions between the endothelial cells of the cerebral capillaries (Fig. 16-3) form the blood-brain barrier. This is why brain ECF has a PCO2 that follows changes in the arterial blood (PaCO2), and a [H+] that follows changes in CSF through the thin pia layer (Fig. 16-3). In metabolic acid-base disorders, the maintained changes in brain ECF [H+] are much smaller than the changes in blood [H+]. The blood-brain barrier has slow transport proteins that can move ions in and out of ECF (Fig. 16-3). Control molecules in area striata, which has widely fenestrated capillaries and no blood-brain barrier, can pass the blood-brain barrier. Breath-holding and hypoventilation leads to carbon dioxide accumulation with acute increase of PaCO2.
Fig. 16-3: The blood brain barrier and the medullary chemoreceptors. Since CO2 is lipid soluble it diffuses rapidly across the blood-brain barrier and increases brain ECF [H+] by hydration to carbonic acid followed by dissociation. Not only this H+ but also PCO2 itself stimulate the medullary chemoreceptors. The sensitivity (gain) of the medullary chemoreceptors to changes in [H+] exceeds that of the carotid chemoreceptors. Most of the steady-state ventilatory rise in response to CO2 is mediated by the medullary receptors, whereas fast and transient changes are first detected by the carotid chemoreceptors. The quantitative relationship between the ventilation and PACO2 is called the CO2 response curve (Fig. 16-4). The slope of such a curve shows the sensitivity or the gain of the RC. At low PACO2-values a small rise does not change the ventilation (see the typical hockey stick curve in Fig. 16-4A). This is because the threshold of the RC has not been reached. The right curve in panel A shows the rise in ventilation by hypercapnia at normal PAO2 (110 mmHg). The left curve shows the response at PAO2 5 kPa (37 mmHg). The slope is steeper which implies an increased sensitivity or gain of the chemosensitive feedback loop during hypoxia. The hypoxia also lowers the threshold for the CO2 stimulus (Fig. 16-4A).
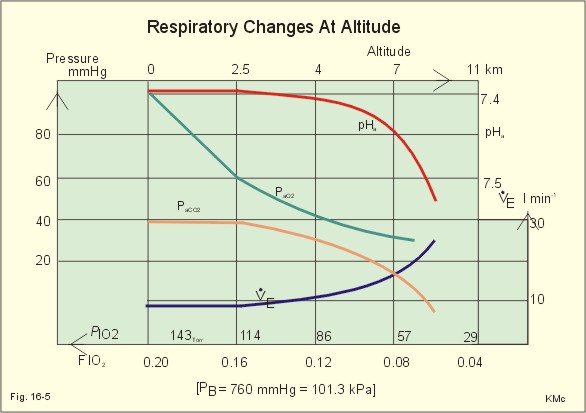
Fig. 16-4: CO2 response curves: A shows results from persons exposed to a combination of hypoxia and hypercapnia, B shows the change from a normal control curve to the response of acidotic patients; C shows the change from control to steady state exercise. The combined effect is multiplicative. The shift to the left of the curve is called reduced threshold due to the new stimulus, which is hypoxia (Fig. 16-4A). The steeper slope of the hockey stick curve is typical for high altitude Acclimatization. The right curve in panel B is a normal control: A low pHa as seen in chronic, metabolic acidosis triggers hyperventilation, so the PACO2 falls further. This is a reduction of the threshold, but not of the sensitivity to PaCO2 of the chemoreceptors. A high blood [H+] is the new stimulus for the acidotic patients of Fig. 16-4B. Exercise has a similar effect (Fig. 16-4C). The CO2 response curve is also shifted to the left, again without a change in the slope. Here, the extra stimuli are signals from the working muscles and from CNS. Acclimatization is the physiologic adaptation of long lasting or repeated stress in the environment . An example of acclimatization or adaptation is observed in inhabitants exposed to chronic hypoxia at high altitudes. Another example is physical training. Acute Acclimatization: On arrival to high altitude a subject experiences immediate hyperventilation. If he becomes a permanent high altitude resident the ventilation gradually decreases a little, as the increase in RBC's compensates for an efficient oxygen transfer. The low PaO2 stimulates the carotid chemoreceptors, causing the rise in ventilation. Acute ventilatory adaptation has an effect upon the peripheral chemoreceptors. The ventilatory response implies excess clearance of CO2 resulting in a reduced PaCO2 and acute respiratory alkalosis, according to the alveolar ventilation equation. The PIO2 in the moist tracheal air will fall with falling FIO2 or with falling atmospheric pressure (PB) at altitude (Fig. 16-5). Ventilation starts to rise at PIO2 values below 13.3 kPa (100 mmHg), which corresponds to a PaO2 of 7.5 kPa, and the rise is hyperbolic (Fig. 16-5). The rise in ventilation in itself reduces the permanent PaCO2 stimulus (Fig. 16-5). The data confirms the presence of an acute respiratory alkalosis with a high pH (Fig. 16-5 and Chapter 17). Chronic Acclimatization: The low PaCO2 triggers adaptive mechanisms in the renal tubule cells (Chapter 25). The renal compensation consists of reduced tubular H+ secretion and thus reduced tubular bicarbonate reabsorption. This in turn then elicits a fall in arterial and CSF [bicarbonate] and a rise of the low [H+]. In the 2-3 days until acute acid-base adaptation is fully accomplished, the arterial [bicarbonate] is reduced in proportion to the fall in PaCO2. Thus pHa is back to normal and the final condition is called compensated respiratory alkalosis (ie, normal pHa and reduced PaCO2). High altitude residents require their own acid-base correction charts with a modified Van Slyke Equation, as using the sea level tables is an attempt to "adapt" them to a different environment and is counterproductive in intensive care. The CSF contains fewer buffers and lower buffer concentrations than the blood. High altitude Acclimatization shifts the CO2 response curve to the left (i.e., reduced threshold), and the slope becomes steeper illustrating a higher sensitivity or gain (Fig. 16-4A). The acclimatized person has similarities to the well-trained athlete with high oxygen capacity (high haematocrit, haemoglobin and erythrocyte count - also similar to chronic polycythaemia). The adaptation is associated with a rise in the circulatory oxygen transport, because both cardiac output and the oxygen capacity of the blood are increased. The erythropoietin release is increased which in turn increases the number of RBC's and gradually reduces the increased ventilation and cardiac frequency, typical of acute exposure to high altitude.
Fig. 16-5: Respiratory variables and their changes at altitude. The acclimatized person has a large blood and plasma volume, growth of the pulmonary arterial wall and the right ventricular muscle mass in response to hypoxic pulmonary vasoconstriction. There is a constant high sympathetic tone, which decreases the renal bloodflow (RBF). In pulmonary disease during chronic hypoxia, the hypoxic vasoconstriction leads to pulmonary hypertension, and right ventricular failure (cor pulmonale) can develop as in chronic mountain sickness now called polyerythrocythemia. During climbing at high altitudes, the low atmospheric pressure (PB) implies a fall in PIO2 =(PB - 6.3)* FIO2 and thus in PaO2. When PaO2 is below 7.3 kPa (55 mmHg), the ventilation must increase progressively, whereby PaCO2 is reduced. The simplified alveolar gas equation (Eq. 14-3) can be applied. The solution for PACO2 is 6 kPa or 45 mmHg. This theoretical calculation of PACO2 is only mathematically correct. The argument is clearly wrong, when PAO2 is below 7.3 kPa, because at this level the hyperventilation will diminish PACO2. Ventilatory Acclimatization elicits a long lasting rise in BTPS-ventilation (measured at altitude), inversely proportional to the fall in PB. A doubling of the BTPS-ventilation at half an atmosphere implies that the ventilation measured in STPD-units is unchanged. According to the alveolar ventilation equation, FACO2 is equal to the ratio between carbon dioxide output and alveolar ventilation (both measured at STPD). Since both of these volumes are unchanged after total adaptation, it follows that also FACO2 must be unchanged. Hence, PACO2 must fall proportional to the fall in PB. However, at the top of Mt. Everest the PB is only 253 mmHg or 1/3 atm, but the ventilation is 5-fold increased. Following return to sea level a fall in ventilation and heart rate takes place over the next three weeks. Also the blood pressure and pulmonary vascular resistance fall druing this period. Adaptation to high altitude and to physical training is lost within a few weeks, depending on the altitude. for example returning from 3600 m at the altitude of La Paz, Bolivia, it takes 20 days to return to sea level values.
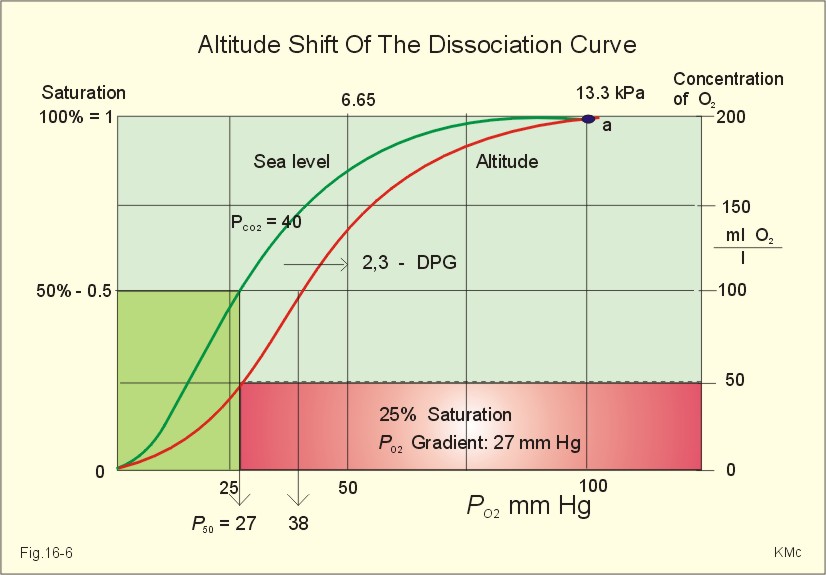
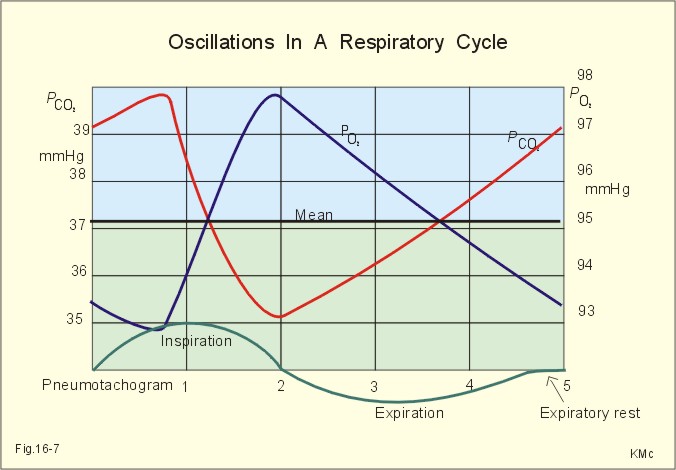
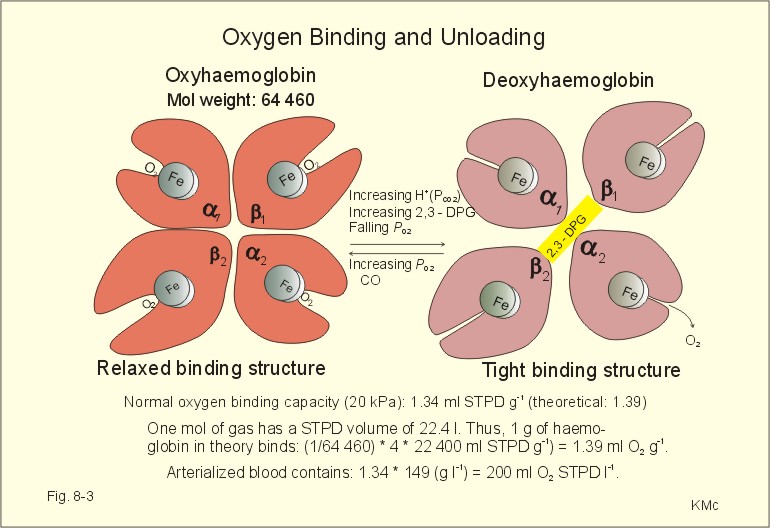
Fig. 16-6: The oxyhaemoglobin curve is shifted towards the right by increased 2,3-DPG content of the red cells. Altitude residents are exposed to chronic hypoxia. They also have high concentrations of 2,3-DPG in their red cells just as endurance athletes, and their oxyhaemoglobin dissociation curve is shifted towards the right (Fig. 8-3 and 16-6). This phenomenon is also expressed by the term P50, which is the PO2 at 50% saturation. The P50 denotes the standard affinity (Ch. 14) between oxygen and haemoglobin. The standard affinity is high, when P50 is low. Normally, the P50 is 3.6 kPa or 27 mmHg (Fig. 16-6). With increased P50 at altitude, the standard affinity is reduced, and the delivery of oxygen to the tissues is improved at a given oxygen concentration. Blood with only 25% saturation has a PO2 of 27 mmHg in altitude residents, much larger than in a sea level resident (see the curve for PCO2= 40 mmHg in Fig. 16-6). 4. Alveolar gas tension oscillations During a normal respiratory cycle the alveolar gas tensions oscillate (Fig. 16-7). During the first part of the inspiration we re-inhale alveolar air contained in the dead space, but soon the tension curves reverse as fresh air is received from the outside. Early in expiration the PCO2 starts to increase linearly and the PO2 falls. These oscillations around the mean alveolar gas tensions are transferred to the passing blood. The carotid bodies may be sensitive to PCO2 oscillations of the arterialized blood.
Fig. 16-7: Oscillations around the mean of the alveolar gas tensions during a normal respiratory cycle performed by a sitting healthy person at rest. The alveolar and arterial oscillations increase in amplitude and frequency during hypoxia and during exercise with tachypnoea and an increased tidal volume. This occurs at high altitude and in all disorders asoociated with hypoxia. Accordingly, any stimulus of the carotid bodies by this mechanism must be accentuated. 5. Oxygen - Altitude - Mt Everest At sea level the ambient or barometric pressure is around 101.3 kPa dependent upon the meteorological status. Above sea level, the ambient pressure decreases with altitude (Fig. 16-8). The top of Mt. Everest is depicted with mountaineer camps for the last 3 km. At any given location on earth, the ambient pressure (PB) equals the gravitational force of the air column per area unit. Classically, PB is given as the height of the mercury (Hg) column, which exerts the same pressure as the air on an area unit (sea level mean 760 mmHg or 101.3 kPa at 0oC and 45o latitude).
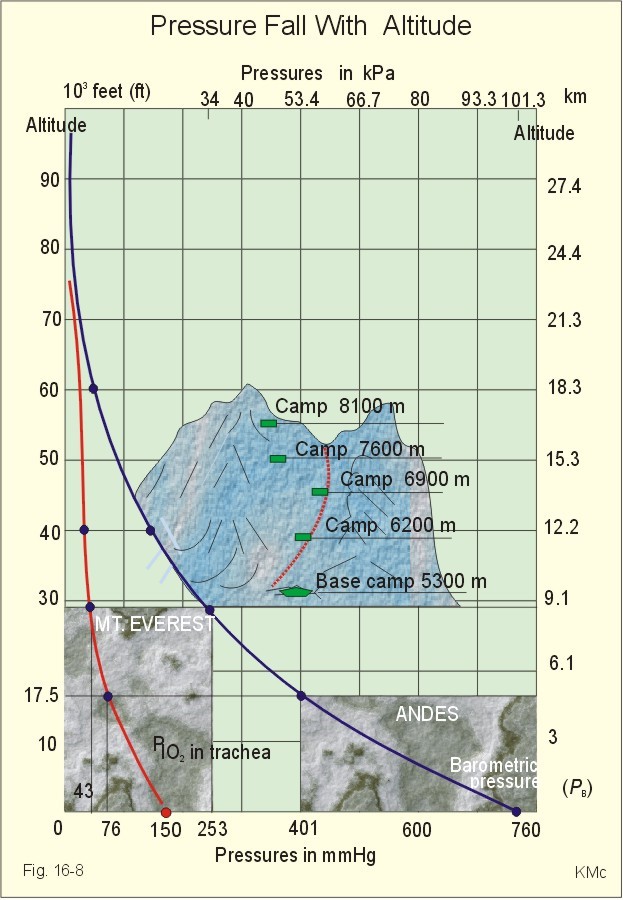
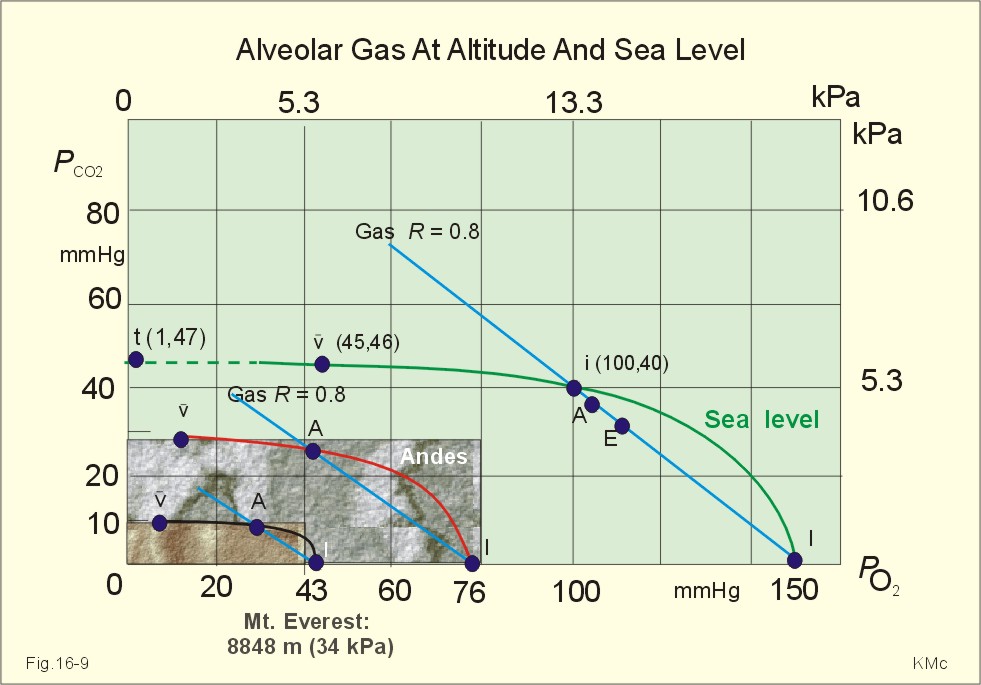
Fig. 16-8: The decrease in inspired oxygen tension (PIO2) and in barometric pressure with increasing altitude at 45 degrees latitude. Since oxygen comprises 20.93% of the air, its partial pressure in moist tracheal air at sea level (water vapor tension 47 mmHg at 37oC) is easily calculated. The molar fraction is equal to the partial pressure of oxygen in the tracheal, moist, inspired air (PIO2) divided by the maximally possible oxygen pressure in this air: PIO2/(760-47) = 0.2093. Accordingly, PIO2 is 150 mmHg or (150* 0.1333)= 20 kPa at sea level. The curve for tracheal PIO2 in Fig. 16-8 is constructed on the basis of such calculations. The alveolar gas equation at an RQ of one, states that PAO2= PIO2 - PACO2 (Eq. 14-3). At a PAO2 of 30 mmHg, most persons lose consciousness, so in theory the lowest critical value of PIO2 is: (30 + 40) = 70 mmHg, with a PACO2 of 40 mmHg. However, a hyperventilation drop in PACO2 leads to a similar rise in PAO2, according to the equation above. Therefore, humans can reach the mountain summits without the use of supplementary oxygen (see below). Altitudes up to 3000 m do not have important adverse effects. The residents of Mexico City live at an altitude of 2200 m without hypoxic problems in general. At an altitude of 3600 m (~11800 ft), tracheal PIO2 is 94 mmHg and, with PACO2 =30 mmHg, PAO2 is expected to be 60 mmHg, as long as the person does not hyperventilate and keep RQ = R =1. In reality, at 3600m, the RQ is .9 slightly higher than at sea level. Such a low PAO2 is an adequate stimulus to the peripheral chemoreceptors (carotid bodies) and ventilation increases. The accompanying drop in PACO2 allows the mountain climber to reach higher and higher altitudes. Residents living in the Andes at an altitude corresponding to a PB of 401 mmHg, inspire air with a tracheal PIO2 of 76 mmHg. This altitude is approximately 17500 feet or 5330 m. Mean values of their alveolar gas composition are PAO2 42, and PACO2 26 mmHg, due to chronic hyperventilation. With an RQ of 0.8 they must have a gas R-value of the same size in order to be in respiratory steady state (Fig.16-9).
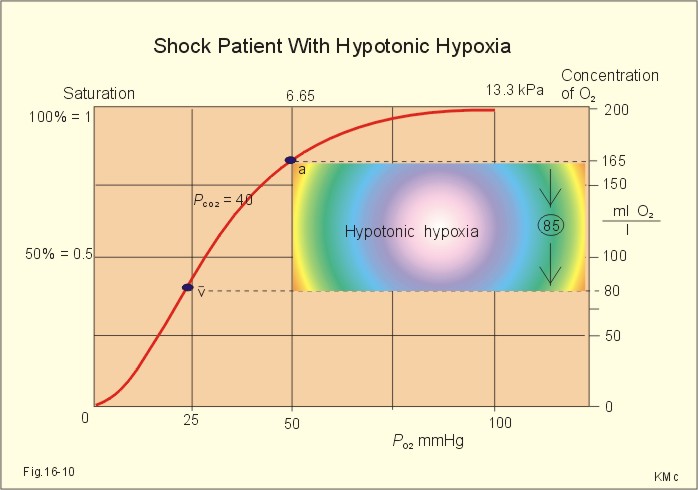
Fig. 16-9: Alveolar points for altitude residents in the Andes, for mountain climbers on Mt. Everest, and for sea level residents. The normal ventilation-perfusion ratio curve for sea level residents is shown with green, the same curve for altitude residents in Andes is shown with red, and the black curve is for climbers on the top of Mount Everest. Depending upon their alveolar ventilation the A point moves up and down the R=0.8 diagonal around the value shown in Fig. 16-9. Altitude residents normally have a small alveolo-arterial PO2 difference. Mt. Everest is located 8848 m above sea level corresponding to a barometric pressure of 34 kPa or 253 mmHg. A Norwegian group of mountain climbers recorded the pressure to 250 mmHg on the top, when setting the height record of 8848 m (1985). The partial pressure of oxygen in the humidified tracheal air is thus: (253-47)* 0.2093 = 43 mmHg or 5.75 kPa (43 in Fig. 16-9). How is it possible to ascent to such a summit, without the use of supplementary oxygen? The A point and the ventilation-perfusion-curve is shown in Fig. 16-9 both for sea level, the Andes and Mt. Everest. A calculation of alveolar gas tension can be performed with the simplified alveolar gas equation (Eq. 14-3). The low oxygen is a strong stimulus to ventilation, and the hyperventilation reduces PACO2 of the acclimatized mountaineer to an average of 8 mmHg or 1 kPa. Accordingly, PAO2 is 35 mmHg (4.67 kPa) or less - close to the critical level of consciousness of 30 mmHg (4 kPa) and the mission is accomplished with an extremely small margin. One member of the above mentioned Norwegian group of mountaineers had amnesia for the last 50 m to the summit and down, although he took pictures from the top together with the group; he remembered only the last part of the climb back to camp. As a matter of fact, a similar simplified calculation seems to predict that the highest mountain to which it is possible to ascend without the use of oxygen does not exist. Let us assume that a PAO2 of 30 mmHg and a PACO2 of 8 mmHg defines the critical barometric pressure. Then Eq 14-3 predicts that 38 mmHg is equal to 20.93% of (PB - 47) mmHg. Accordingly, the critical PB is (182 + 47) = 229 mmHg, corresponding to more than 9 km of altitude. Such a postulate can never be proven in real life, because Mt Everest, with 8848 m, is the highest mountain on Earth. The simplified calculations are rough estimates, because the critical conscious level of 30 mmHg is in the arterial blood - not in the alveolar air. The prediction has recently been tested in hypobaric chambers, where indians from the Andes remained well at a pressure corresponding to more than 9 km of altitude. The adaptation to high altitude can even be to the hypoxic levels of the summit of Mt. Everest, as postulated by Prof. Dr. Gustavo Zubieta-Castillo(Sr,). This includes a very slow ascent, protection from cold weather and adequate nutrition. This paragraph covers 1. Shock, 2. Hypermetabolism, 3. Chronic anaemia and 4. Mountain sickness. Patients with falling cardiac output and imminent shock, are forced to increase their arterio-venous oxygen content difference (ie, tissue extraction), since the necessary cellular O2 uptake does not fall appreciably in the case shown in Fig. 16-10. The CAO2 is 165 and the concentration in the mixed venous blood is 80 ml*l-1 (assuming a normal haemoglobin concentration). Thus, the normal resting oxygen content difference of 50 is increased to a tissue extraction of 85 ml*l-1 (Fig.16-10).
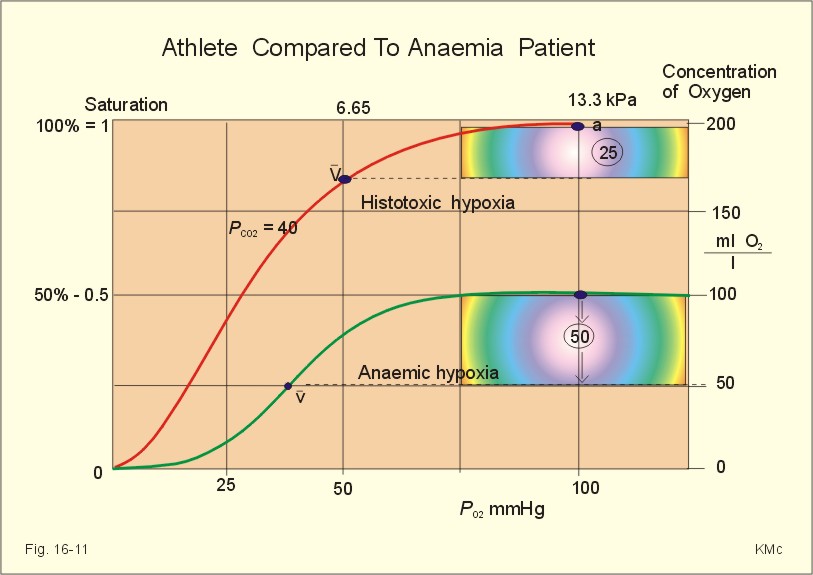
Fig. 16-10: The arterial (a) and mixed venous point on the oxyhaemoglobin saturation curve of a shock patient with falling cardiac output. For other aspects of shock - see Chapter 12. Exhausted athletes may develop hypermetabolism, where ATP degradation exceeds the production due to decoupling of ATP synthetase, whereby heat is released instead of ATP in the respiratory chain. The decoupling is perhaps due to a thermal change in the protein configuration of ATP synthetase. These athletes are unable to utilize the high oxygen tension (Fig. 16-11). The tissue extraction of oxygen is insufficient - only 25 ml l-1 (Fig. 16-11).
Fig. 16-11: Arterial (a) and mixed venous (v) oxygen tensions on the oxyhaemoglobin dissociation curve of an athlete with hypermetabolism, and of a severely anaemic patient. Chronic anaemia may develop so slowly that the patient can still work, although deprived of half the haemoglobin content of the blood (Fig. 16-11). The arterio-venous oxygen content difference is normal (50 ml l-1 in Fig. 16-11), and the venous oxygen tension gradient, responsible for delivery of oxygen to the most hypoxic tissues, is relatively high. Patients with anaemia, chronic pulmonary disease and cyanotic heart disease, have high erythrocyte concentrations of 2,3-DPG (see Fig. 8-3). This molecule shifts the oxyhaemoglobin dissociation curve to the right, just as it occurs in hypercapnia and at increased body temperature. The 2,3-DPG diminishes the haemoglobin affinity for oxygen, so the binding in the lungs is impaired, while the peripheral release of oxygen is facilitated. The effect is beneficial for the tissue oxygenation of the anaemic patient, because of a larger PO2 gradient at a given blood oxygen content. This concept covers several disorders, which are divided into acute mountain sickness (AMS) and chronic mountain sickness (CMS). Acute Mountain Sickness Acute Mountain Sickness has 1. A frequent benign and 2. A rare malign form. 1. Benignant Acute Mountain Sickness hits up to 40% of the 30 million persons yearly attracted by the high altitude environment above 3500 m. Symptoms occur within 6-12 hours of arrival and include frontal headache, malaise, nausea, lassitude and insomnia - mainly cerebral symptoms. The normal response of a healthy person to hypobaric hypoxia is a rise in ventilation within seconds, and a rise in urine flow developing over 10-20 min. The hypoxic pulmonary vasoconstriction increases the right atrial pressure and releases the hormone, atrial natriuretic peptide, from the atrial walls to the blood. Atrial natriuretic peptide increases the excretion of Na+ and water in the urine. Persons with high normal blood levels of the hormone are less prone to develop acute mountain sickness. The typical patient reacts – at the time the disease develops - with a relatively small hyperventilation due to hyposensitive carotid bodies. The drop in oxygen tension does not affect the carotid bodies in these patients as much as in healthy persons. The PaCO2 is not reduced as much as in the healthy subject. The patient secretes more H+ and reabsorbs more bicarbonate and Na+ than normal. This results in Na+ retention and in water accumulation. The relatively high PaCO2 also leads to a relatively high cerebral bloodflow. This is in contrast to the healthy mountaineer who has a relatively low PaCO2, which causes less cerebral vasodilatation and less cerebral disorder. Slow Acclimatization by slow ascent is the best prophylactic advice to mountaineers. Since part of the mechanism of acute mountain sickness is obviously related to the degree of respiratory alkalosis, it is no surprise that prophylactic treatment with carboanhydrase inhibitors (eg, acetazolamide) reduces the symptoms, because of the increased bicarbonate elimination in the kidneys, and because of the increased tissue PCO2, which stimulates ventilation (“artificial Acclimatization”). 2. Malignant Acute Mountain Sickness includes high-altitude pulmonary oedema (HAPE) and high-altitude cerebral oedema (HACE). They are rather infrequent, life-threatening forms with an incidence of 1% among mountaineers. HAPE is established when a dyspnoeic subject has bloodstained frothy sputum or substantial chest rales. The salt- and water-accumulation increases the haemodynamic pressure and reduces the colloid-osmotic pressure by dilution. Both changes tend to produce oedema. This is a condition of imminent death from cardiovascular and respiratory failure, unless treated rapidly. HACE is established when a drowsy mountaineer develops ataxia, nystagmus, papiloedema (retina is brain tissue) and coma. This condition is probably caused by extreme salt and water retention. HAPE and HACE must be treated urgently with oxygen and descent to a lower altitude as fast as possible. The glucocorticoids, dexamethasone and betamethasone, exhibit minimal mineralocorticoid effect, maximal anti-inflammatory activity. These drugs are the choice in case of acute brain and lung oedema. The adaptation to hypoxia is classified as Acute, Subacute and Chronic. It is best judged by the increase of the number of red blood cells. When the red cell count increase reaches a plateau full adaptation to hypoxia is achieved. Notice the three sections where Acute, Subacute and Chronic Mountain Sickness can occur in the susceptible individuals and those with lung or heart disease (Fig. 16-12). Notice also that a subject with a normal Hematocrit of 36 % at sea level can increase to 50% at 3600 m. The subject can oscillate between these values if he changes residence at least every month and a half. The time for full adaptation to high altitude is calculated using the high altitude adaptation formula (Eq. 16-1). Going from sea level, the altitude in Km is multiplied by 11.4 days in order to calculate (with this simplified formula) when the approximate full adaptation to high altitude takes place.
Fig. 16-12: The three stages of adaptation to high altitude, from sea level to 3600m.
Chronic Mountain Sickness (CMS) Chronic Mountain Sickness can occur in some long-term residents of altitudes above 2500 m - the Andes and elsewhere. The number of red cells in the blood (polycythaemia) and the haematocrit develops to exceptionally high values. The high haematocrit and viscosity of the blood do not necessarily lead towards the formation of emboli, which occurs when there is concurrent phlebitis or other vascular disease. In patients with lung disease, pulmonary vasoconstriction maintained over years can lead to pulmonary hypertension and enlargement of the right ventricle, resulting in congestive heart failure of the right heart - Latin: cor pulmonale (see Chapter 10 and 14). A large fraction of the cardiac output is shunted through vessels without alveoli or vessels with hypoventilated alveoli, so the patient is cyanotic with congested ear lobes and some with finger clubbing. CMS is an inadequate denomination for pulmonary disease at high altitude, associated frequently to pulmonary shunts with increased erythropoyetin and polyerythrocythemia (from the Greek: poly = increase, erythro = red, cyt = cells, hemia = in blood). The term previously used, CMS should be discarded and polyerythrocythemia be used instead as a sign of lung disease at high altitude. Furthermore the concept of "Loss of Adaptation" was incorrect since it was a mere attempt to avoid the explanation of the ethiopathophysiology of the disease. Quoting Prof. Zubieta-Castillo (Sr): “The organic systems of human beings and all other species tend to adapt to any environmental change and circumstance, and never tend towards regression which would inevitably lead to death". It is important to diagnose in these patients at high altitude if they are suffering from the triple hypoxia syndrome (THS). THS is the superposition of three hypoxias: 1) First hypoxia: high altitude, 2) Second hypoxia: polyerythrocythemia or chronic lung disease hypoxia and 3) Third hypoxia: Acute superimposed lung disease such as the flu, interstitical edema or pneumonia.
Fig. 16-13: The Triple Hypoxia Syndrome at high altitude, an essential tool for the diagnosis of patients suffering from polyerythrocythemia. The importance underlies in the fact that the third hypoxia is reversible and treatable. Futhermore, many patients with what was previously know as CMS, consulted during a THS episode and hence created confusion in the interpretation of the disease. The treatment of polyerythrocythemia is improvement and control of lung function at high altitude. Some cases can descend to sea level, however the overlying disease has to be treated, prior to departure.
Equations - see Chapter 14. · The alveolar gas equation · The alveolar ventilation equation · The final ventilation-perfusion-equation The high altitude adaptation formula going from sea level to a fixed altitude is: : I. Each of the following five answers have True/False options: Statement: The hyperventilatory response to hypoxia and hypoxaemia is mediated by the: A. Bronchopulmonary mechanoreceptors B. Chemoreceptors in the carotid bodies C. Central chemoreceptors D. Irritant airway receptors E. Arterial baroreceptors. II. Each of the following five statements have True/False options: A. The primary muscle of inspiration is the diaphragm. B. The airflow resistance is highest in the small terminal bronchioles. C. The intrathoracic pressure is less than atmospheric pressure. D. The VC is defined as the maximum volume of air that can be inhaled following a maximum expiration. E. The lung compliance is reduced in persons with emphysema. III. Each of the following five statements have True/False options: A. Polycythaemia patients have lots of red cells and a large oxygen binding capacity ([Total-Hb] * 1.34). B. Patients with a high haemoglobin concentration show cyanosis as frequent as do patients with anaemia. C. The typical patient with acute mountain sickness reacts with a high diuresis. D. The 60 g of reduced haemoglobin in one litre of average capillary blood is the approximate threshold for the bedside diagnosis of visible cyanosis. E. Patients with anaemia, chronic pulmonary disease and cyanotic heart disease, have high erythrocyte concentrations of 2,3-diphosphoglycerate (2,3-DPG). A healthy female, with an anatomic VD of 0.12 l and a respiratory frequency (f) of 14, is flying in an open-cockpit aeroplane at 2000 m. Here, her PIO2 is 16 kPa (120 mmHg) and her alveolar ventilation is increased to 5.6 l STPD per min (from 4.2 at the ground). She has had a mixed diet meal before take-off, and her R- value remains at 0.8. Her PAO2 is restored to 13.3 kPa (100 mmHg) by the rise in ventilation. 1. Calculate her PACO2 by the simplified alveolar air equation. 2. Calculate her expired minute ventilation. A group of altitude residents (Sherpas) reside for a month at an altitude of 6 km with a PB of 410 mmHg (5.47 kPa). After three weeks their average alveolar gas tensions were PAO2 40 (5.3 kPa) and PACO2 22 mmHg (2.9 kPa). The average metabolic rate at rest was 5000 J per min or 84 Watts. The respiratory energy equivalent for oxygen on a mixed diet is 20 kJ per l STPD. A PAO2 of 30 mmHg (4 kPa) is the threshold for loss of consciousness. 1. Calculate the alveolar minute ventilation in STPD volume units and at the altitude (in l BTPS). 2. Calculate the lowest barometric pressure (PB) at which man can survive by breathing air. Assume that PACO2 is 8 mmHg (1 kPa) at the summit. A polio patient, 11 years old (body weight 35 kg), is connected to a pressure-controlled respirator, because the boys respiratory muscles are paralyzed. He receives atmospheric air through a tracheal tube by positive-pressure ventilation (input pressure 800 Pascal, Pa) at a frequency (f) of 10 per min. The equipment dead space and the dead space of the small boy added together amounts to 130 ml BTPS. At FRC the specific lung compliance is 1.5 ml BTPS per Pa, and the specific compliance of the thoracic cage is 0.8 ml BTPS per Pa. The specific total standard compliance of a healthy adult is 1 ml BTPS per Pa. The barometric pressure (PB) is 770 mmHg (102.64 kPa), and the FACO2 is 0.059. The arteriovenous oxygen content difference is 55 ml STPD l-1. The patient is in respiratory steady state with a respiratory exchange quotient of 1. 1. Calculate the specific total standard compliance of the polio patient. 2. Calculate the size of the alveolar ventilation. 3. Calculate the carbon dioxide output in ml STPD. 4. Calculate the cardiac output. Try to solve the problems before looking up the answers. · Medullary chemoreceptors communicate with the respiratory centre and respond to acidaemia or hypercapnia with increased ventilation. · The glomus cells of the carotid body respond to hypoxaemia, hypercapnia, acidaemia, and hyperkalaemia with increased ventilation. · Chronic respiratory acidosis is compensated renally by bicarbonate reabsorption. · Respiratory insufficiency in chronic obstructive lung disease is endangered when breathing pure oxygen. · Chronic hypoxia adaptation is by increased erythropoiesis, increased 2,3-DPG production, and reduced ventilatory response to acute hypoxia. · Ventilatory altitude Acclimatization is a hypoxic chemoreflex. · The respiratory altitude alkalosis is compensated renally in days. · Altitude Acclimatization is similar to hypoxia-training and endurance training. The high altitude adaptation formula allows for a precise calculation of full adaptation to chronic hypoxia from the hematologic point of view. · The normal response of a healthy person to hypobaric hypoxia is a rise in ventilation within seconds, and a rise in diuresis developing over 10-20 min. The hypoxic pulmonary vasoconstriction increases the right atrial pressure and releases the hormone, atrial natriuretic peptide from the atrial walls to the blood. Atrial natriuretic peptide increases the excretion of Na+ and water in the urine. · Persons with high normal blood levels of atrial natriuretic peptide are less prone to develop acute mountain sickness. · The typical mountain sick patient reacts with a relatively small hyperventilation due to hyposensitive carotid bodies. The fall in oxygen tension does not affect the carotid bodies in these patients as much as in healthy persons. The PaCO2 is not reduced as much as in the healthy subject. The patient secretes more H+ and reabsorbs more bicarbonate and Na+ than the normal. This results in Na+ retention and in water accumulation. The relatively high PaCO2 also leads to a relatively high cerebral bloodflow. · This is in contrast to the healthy mountaineer who has a relatively low PaCO2 , which causes less cerebral vasodilatation and less cerebral disorder. · Chronic Mountain Sickness (a deprecated term) occurs in long-term residents of altitudes above 2500 m - the Andes and elsewhere. It is now called polyerythrocithemia where the number of red cells in the blood and the haematocrit develops to exceptionally high values. · Pulmonary vasoconstriction maintained over years leads to pulmonary hypertension and enlargement of the right ventricle, resulting in congestive heart failure of the right heart - Latin: cor pulmonale. Gonzales C, L Almaras, A Obeso, and R Rigual (1994). Carotid body chemoreceptors; From natural stimuli to sensory discharges. Physiol. Rev. 74: 829-898. Hultgren, HN (1997) High altitude medicine. Stanford, California. Zubieta-Calleja, G, Zubieta-Castillo G. (1994) High altitude pathology at 12000 ft. La Paz, Bolivia http://www.altitudeclinic.com/pub
|
||





.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Click here to introduce your comments or contributions